Mustguseal Protocol
Mustguseal aims at constructing large alignments of functionally diverse protein families by automatically collecting the available information about their structures and sequences in public databases. To meet this objective the Mustguseal protocol implements a combination of structure/sequence search/comparison algorithms to take into account sequence and structural variability of functionally diverse homologs within a large superfamily. All bioinformatic routines are fully automated and executed entirely on the server side. This page gives an overview of the Mustguseal protocol. The detailed description of the protocol is provided in the Mustguseal publication.
 Click to open in full-screen mode
Click to open in full-screen mode
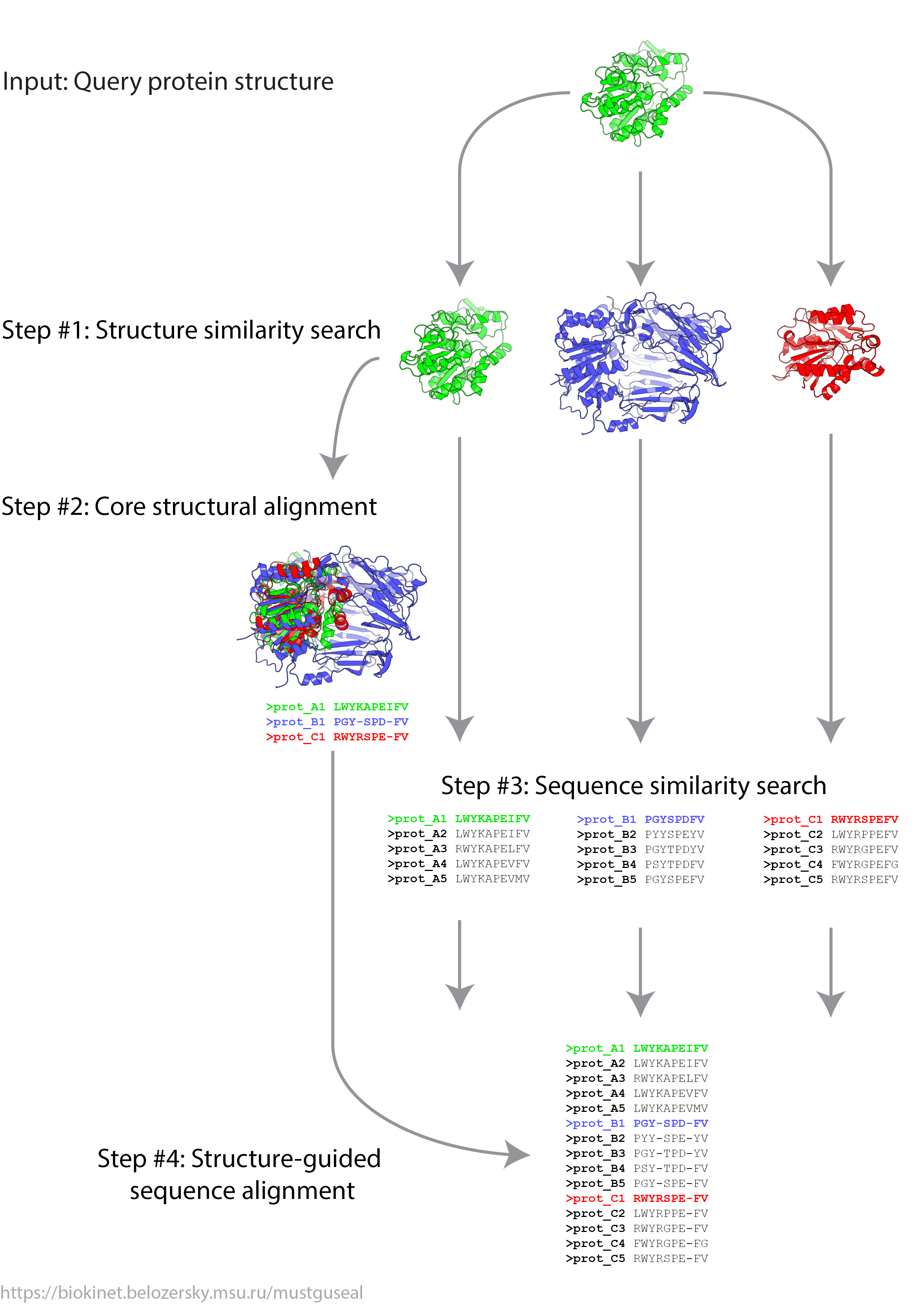
The Mustguseal protocol contains four major steps:
- Step 1: Structure similarity search
- Step 2: Construction of the core structural alignment
- Step 3: Sequence similarity search
- Step 4: Structure-guided sequence alignment
Step 1: Structure similarity search The default input to the server (i.e., in Mode 1) is a protein structure submitted as PDB ID and chain ID. Protein structures are considered to be more conserved throughout the evolution compared to sequences. Therefore, structure similarity search by the SSM algorithm versus the entire PDB database is used to collect evolutionarily remote relatives of the query protein. These distant homologs share a common structural framework but lost sequence similarity during natural selection and evolution from a common ancestor, and thus are likely to have broad functional variability. The collected set of proteins is expected to represent different protein families with various functions within a superfamily, and will be further referred to as the representative set.
Step 2: Construction of the core structural alignment The representative set of proteins is aligned using the MATT multiple structural alignment program. The structural alignment of a representative set of homologous proteins is the core of the Multiple Structure-Guided Sequence Alignment which defines its scope and diversity, and will be further referred to as the core structural alignment. It is important that proteins in this core structural alignment represent the desired diversity among the protein families of interest. When using the Mode 1 (i.e., the representative set of proteins is collected and aligned automatically) users are advised to download this superimposition using a link at the Results page and evaluate: (1) if the automatically selected proteins represent the desired diversity among the protein families of your interest, and (2) if the automatically created structural alignment is accurate (special attention should be paid to flexible loop regions and crucial non-standard/modified amino acids). A user-defined/edited core structural alignment can be submitted as a new task in Mode 2 or Mode 3. See more information on the input modes in a corresponding section of the on-line manual
Step 3: Sequence similarity search Each protein from the core structural alignment, i.e., each representative protein, is independently used as a query to execute BLAST sequence similarity search and collect evolutionarily close relatives - members of the represented families. Filters are applied to eliminate redundant entries as well as too distant proteins (i.e., outliers) within each group using the CD-HIT and HHFilter routines. Sequences within each group are aligned by implementing the MAFFT multiple sequence alignment algorithm.
Step 4: Structure-guided sequence alignment The alignment of sequences of the evolutionarily distant relatives is created by using the structural alignment of the representative proteins as a guide. During this step the superimpositions built on Steps 2 and 3 by MATT and MAFFT do not change. Columns of gaps are inserted into individual sequence alignments in a way that their total lengths become equal and the superposition of the representative proteins in the merged sequence alignments matches their superimposition in the core structural alignment.