parMATT
Parallel multiple alignment of protein 3D-structures with translations and twists for distributed-memory multiprocessor systems 
A 3D-alignment of multiple protein structures is fundamentally important for a variety of tasks in modern biology and becomes more time-consuming with the increase of the number of PDB records to be compared. Ten years ago it was common to superimpose just a few protein structures due to a limited amount of 3D-data deposited at that time. Today, non-redundant collections of protein superfamilies are represented by hundreds of 3D-records, making it problematic to use the available single-CPU software to perform such a superimposition. More than 146 million sequence entries of the currently known proteins are deposited in the UniProtKB database, and as the PDB database demonstrates a geometric growth we are facing further increase in the number of known protein structures corresponding to diverse superfamilies, ruling out the use of single-CPU 3D-alignment programs at a daily routine in the future.
The parMATT is a hybrid MPI/pthreads/OpenMP parallel re-implementation of the MATT algorithm designed to benefit from the growing availability of structural data by accelerating multiple structural alignment at large-scale analysis of protein families/superfamilies. The parMATT can be faster than MATT on a single multi-core CPU, and provides a much greater speed-up on distributed-memory systems, i.e., computing clusters and supercomputers hosting memory-independent computing nodes. The parMATT can significantly accelerate the time-consuming process of building a multiple structural alignment from a large collection of 3D-models of homologous proteins. The output of MATT and parMATT are identical.
The parMATT is the first and only program currently available which supports the MPI level of parallelism at aligning multiple protein structures.
Note: Prior to running the parMATT make sure that use set the appropriate output mode depending on your research objective (i.e., “Partial alignments” enabled or disabled) - see this link for details.
THIS WEB-SITE/WEB-SERVER/SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS OR HARDWARE OWNERS OR WEB-SITE/WEB-SERVER/SOFTWARE MAINTEINERS/ADMINISTRATORS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE WEB-SITE/WEB-SERVER/SOFTWARE OR THE USE OR OTHER DEALINGS IN THE WEB-SITE/WEB-SERVER/SOFTWARE. PLEASE NOTE THAT OUR WEBSITE COLLECTS STANDARD APACHE2 LOGS, INCLUDING DATES, TIMES, IP ADDRESSES, AND SPECIFIC WEB ADDRESSES ACCESSED. THIS DATA IS STORED FOR APPROXIMATELY ONE YEAR FOR SECURITY AND ANALYTICAL PURPOSES. YOUR PRIVACY IS IMPORTANT TO US, AND WE USE THIS INFORMATION SOLELY FOR WEBSITE IMPROVEMENT AND PROTECTION AGAINST POTENTIAL THREATS. BY USING OUR SITE, YOU CONSENT TO THIS DATA COLLECTION.
News and updates
[2020-11-05] parMATT v.1.2 released implementing bug fix (see the User's Manual for details)
[2020-04-11] The parMATT manual has been updated (both on-line and PDF versions) to explain the “Partial alignments” postprocessing of the 3D-alignment (view on-line)
[2019-04-14] parMATT v.1.1 released (see the User's Manual for details)
[2019-03-27] The parMATT paper published in Bioinformatics DOI:10.1093/bioinformatics/btz224
Download parMATT
parMATT v. 1.2 [2020-11-05] download
parMATT v. 1.1 [2019-04-14] download
parMATT v. 1.0 [2018-02-20] download
Download accessory scripts
Split parMATT/MATT output PDB file to separate PDB structures
splitMATT2chains.sh v. 1.1 [2020-07-28] download
See the User's Manual for details (chapter "Post processing of the parMATT/MATT’s 3D alignment file"). The example dataset can be downloaded using this link: download.
Convert the common structural core instruction file for JMol into the PyMol format
jmol2pymol.pl v. 1.0 [2018-12-20] download
See the User's Manual for details (chapter "Analysis of the common structural core").
Citing parMATT
If you find parMATT or its results useful please cite our work:
Shegay M., Suplatov D., Popova N., Švedas V., Voevodin Vl. (2019) parMATT: Parallel multiple alignment of protein 3D-structures with translations and twists for distributed-memory systems, Bioinformatics DOI:10.1093/bioinformatics/btz224
The parMATT is based on the MATT algorithm and source code: Menke M., Berger B., and Cowen L. (2008). Matt: local flexibility aids protein multiple structure alignment. PLoS computational biology, 4(1), e10.
The parMATT's User's Manual
The User's Manual provides a detailed description of the parMATT program and its features, including installation and execution syntax. The following chapters are included in the User's Manual:
- Version history
- Prerequisites
- Compilation
- The parMATT's options and variables
- The parMATT's input
- Running parMATT
- The parMATT's output
- parMATT’s output: understanding “Partial alignments”
- The parMATT’s parallel performance and scalability
- Post processing of the parMATT/MATT’s 3D alignment file
- Analysis of the common structural core
- The parMATT's examples
- Collecting a set of 3D-models of homologous proteins
- Implementation of parMATT in the laboratory practice
- The parMATT's license
- Citing parMATT
| Download PDF file (1.7 MB) [version: 2020-11-05] |
The parMATT: In a nutshell
A brief overview of the parMATT is provided below. You should see the User's Manual for a full description of parMATT's features and options.
- Prerequisites
- Running parMATT
- The parMATT's input and output
- parMATT’s output: understanding “Partial alignments”
- The parMATT's examples
- The parMATT's license
[return to toc]
Prerequisites
The parMATT is not hardware-specific and is expected to run on any architecture under a Linux/Unix operating system. The parMATT can be launched on a regular desktop multi-core CPU, but its main advantage is the ability to run on distributed-memory multiprocessor systems, i.e., computing clusters and supercomputers hosting memory-independent computing nodes. Installation of parMATT from sources is straightforward, does not require significant investment of time from the user, and can be performed by free tools (i.e., GNU C++ and MPI compilers). See the User's Manual for details.
[return to toc]
Running parMATT
The parMATT does not have a graphical interface and has to be executed from a command-line. The parMATT software is faster then MATT on a single desktop CPU and can provide much greater acceleration on distributed-memory systems, i.e., computing clusters and supercomputers hosting memory-independent computing nodes. The difference between running MATT on a local computer and running parMATT on a computing cluster/supercomputer is explained below:
- to run on multiple nodes (i.e., multiple CPUs) parMATT has to be launched as an MPI program by the appropriate MPI utility (not required for local execution on a desktop computer);
- the ‘
-t t' parameter should be set equal to the number of physical cores in the CPUs which are used in your computing cluster/supercomputer, i.e., the ‘-t t' parameter sets the numbers of cores to be used on each node, and the number of nodes should be set as a separate parameter to the MPI utility (see an example below).
Exact command to launch parMATT depends on your computing cluster/supercomputer. But once you learn this hardware/software-specific command and the number of physical cores in your CPU model, running parMATT will be as easy as running any other program on your local computer.
Launch parMATT locally on 4 physical cores of a single Desktop CPU:
/path/to/parMatt -t 4 -L input.list -o output
Launch parMATT on 8 nodes (i.e., 8 CPUs), 14 physical cores on each node, using the mpirun:
mpirun -np 8 /path/to/parMatt -t 14 -L input.list -o output
See the User's Manual for details and more examples.
[return to toc]
The parMATT's input and output
The input to parMATT is a set of protein structures in the PDB format. One PDB file should represent a single protein chain.
The primary parMATT's output is (1) a file in the PDB format with 3D superimposition of all input structures, and (2) a file in the FASTA format with a corresponding structure-based sequence alignment of the common structural core (i.e., structural equivalences which are shared by all proteins from the input set). The parMATT inherits the bioinformatics part (i.e., the algorithm), the input and the output formats, options and environmental variables from the MATT source code, and thus the parMATT's output alignment is identical to that of the MATT.
The following default output files are produced by parMATT on successful completion:
- a 3D coordinate representation of a multiple structural alignment, i.e., a PDB file with aligned coordinates of all 3D-models from the input;
- a structure-based sequence alignment of the common core, i.e., a sequence alignment file in FASTA format;
- a text file with a summary of the input PDBs (the pairwise comparison tree) and the output superimposition (number of residues in the core alignment, RMSD of the core alignment, the MATT's alignment quality score and the sequence representation of the common core alignment in the PHYLIP format);
- a Rasmol script to highlight aligned residues.
[return to toc]
parMATT’s output: understanding “Partial alignments”
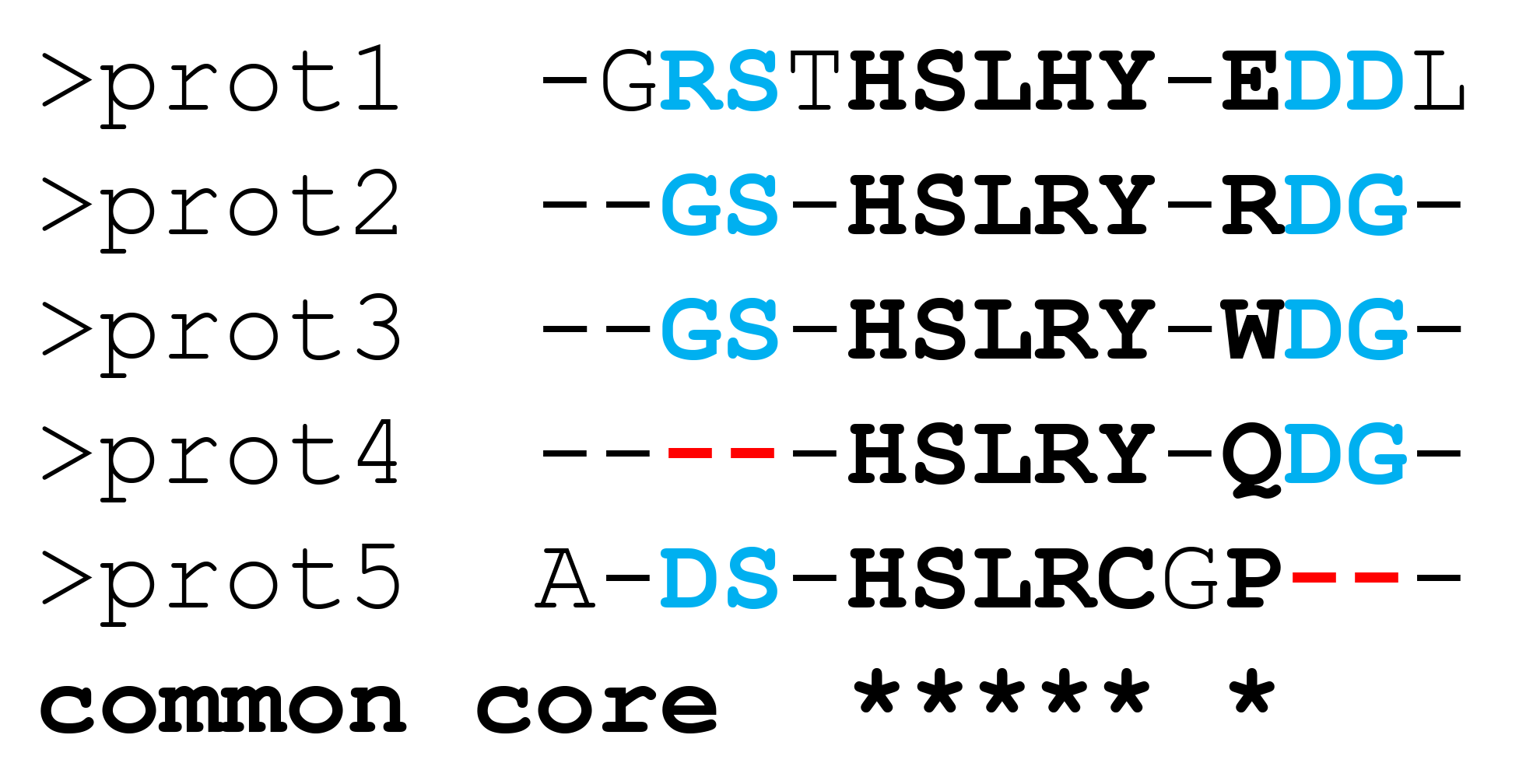
In summary. By default, the produced sequence representation of the constructed 3D-alignment (i.e., the FASTA file) will effectively contain only the "common core" alignment - see the Fig.A below. To further enrich the sequence-based representation of the 3D-alignment by including partially aligned "columns" (i.e., to obtain output similar to Fig.B below) use an additional command line option "-p1" to activate the "Partial alignments" step. This last step is OpenMP only, i.e., it will run in parallel on all threads available on the first node.
To activate the “Partial Alignments” algorithm simply add the "-p1" flag to your command line, e.g., to run locally on 4 CPU cores with “Partial Alignments” you can use the following command:
/path/to/parMatt -t 4 -p1 -L input.list -o output
As explained above, the output of parMATT is the 3D-alignment in the PDB format and its sequence representation in the FASTA format. The conversion of the 3D-alignment (i.e., superimposition of atom coordinates in the 3D-space) into the alphabet form is actually not as trivial as it may seem. In fact, it is complex and highly resource-demanding process.
By default, the parMATT will produce the FASTA alignment of only the common core residues, i.e. structural equivalences which are shared by all proteins from the input set, as shown below on Figure A. This representation may be further improved by implementing the “Partial Alignments” algorithm. Formally, the “Partial Alignments” step is not part of the MATT algorithm and has no effect on the structural superimposition of input proteins (i.e., the output PDB files containing the 3D-alignment will be identical with or without this step). When this step is enabled, the sub-alignments of residues that are not equivalent in all proteins but can be matched in some homologs (i.e., the partial alignments) will be included in the FASTA file to accompany the common core alignment, as shown below on Figure B. This will produce a more content-rich output and facilitate its visual inspection, but will require additional computer time.
To activate the “Partial Alignments” step add the ‘–p1’ flag (no space) to the parMATT command. The default is ‘–p0’.
|
|
|
A. Sequence representation of 3D-alignment with “Partial alignments” disabled |
|
|
|
B. Sequence representation of 3D-alignment with “Partial alignments” enabled |

[return to toc]
The parMATT's examples
Please see the User's Manual for more details regarding the examples. We provide two test datasets for two purposes:
- Example 1 (a small dataset). This dataset contains only 5 protein structures of highly structurally similar MAP kinases. The multiple structural alignment of this set by parMATT should take just a few seconds on any modern hardware. Thus, the purpose of this data set is to quickly verify whether your build of parMATT’s binary from the source code was successful. You can download the dataset, including the input PDB files as well as pre-calculated output files, using this link: [download example 1]. If the program fails to produce the output or takes a significant amount of time on this example you should revise the compilation procedure.
- Example 2 (a large dataset). The purpose of the large dataset (111 protein structures) is to test scalability of parMATT on your multiprocessor system. Before using the parMATT to do actual work you are advised to run the program several times with different resources, i.e., on 1 node/CPU, 2 nodes/CPUs, 4 nodes/CPUs, etc, and evaluate the scalability of your parMATT build on your particular hardware. The exact acceleration will depend on the particular configuration of your multi-processor system. However, if you do not experience any significant speedup at all, that would indicate a problem. You can download the dataset, including the input PDB files as well as pre-calculated output files, using this link: [download example 2].
To evaluate the computational performance of parMATT on distributed-memory multiprocessor systems thirty non-redundant sets of hundreds of protein structures sharing a common structural core and corresponding to the largest CATH superfamilies have been constructed. The protocol which was used to construct the sets is described in the parMATT publication. You can download these test sets using this link: [download parMATT test sets].
[return to toc]
The parMATT's license
The parMatt is licensed under the GNU public license version 2.0.