Mustguseal Output
The output is presented on two pages. The Download section provides links to the primary output - the final alignment - and supplementary (intermediate) output - core structural alignment, structure similarity search results, and sequence similarity search results. The Analysis section offers basic alignment statistics and interactive content for sequence and structure analysis. Interactivity is implemented in HTML5 and therefore no plugins nor Java are required. These tools can help with evaluating the coverage and quality of your alignment and conducting a preliminary study of your protein families. Advanced tools to further study the Mustguseal alignment are discussed in a separate section.
- Download section
- Analysis section: Basic alignment statistics
- Analysis section: Sequence analysis
- Analysis section: Structure-based annotation

The Download section provides links to the Primary output - the final alignment - and the Supplementary output - core structural alignment, structure similarity search results, and sequence similarity search results. All files include the TaskID in their names and have the 'tar.gz' format. In Linux use tar xzf file.tar.gz to extract the data (both tar and gzip usually come with the Linux distribution). In Windows you can use a free tool 7-zip to unpack the archive.
The FINAL_TaskID.tar.gz archive has the following content:
FINAL_TaskID.fasta- the final multiple structure-guided sequence alignment in FASTA format.
The strcore_TaskID.tar.gz archive has the following content:
strcore_TaskID.pdb- multiple 3D-structural alignment of the selected representative proteins in the PDB format. This file contains the "true" 3D-structural alignment, i.e., 3D-entries of all selected proteins superimposed in the 3D-space;strcore_TaskID.fasta- sequence representation (FASTA format) of the core structural alignment of the representative set of proteins;strcore_TaskID.txt- plain text file containing 3D-alignment quality indicators (size of the common 3D-core, its RMSD, and the alignment score according to the MATT algorithm);original_pdbs/- folder with the original (i.e., input) PDB files of the representative proteins;aligned_pdbs/- folder with PDB files of the representative set of proteins which preserve the common coordinate space. I.e., if these files were to be opened at once by PyMol, the viewport would present the 3D-alignment of these PDB entries. Please note, that PDB files in this folder may differ from the original files (i.e., present in theoriginal_pdbs/folder), e.g., heteroatoms will be removed and non-standard formatting may also be lost;corealn_split2pdb.log- log file describing how individual PDB files in thealigned_pdbs/folder were obtained from the all-in-one 3D-alignment filestrcore_TaskID.pdbthat was originally prepared by parMATT/MATT.
The strsearch_TaskID.tar.gz archive has the following content:
superimpose.list- file with the results of the structure similarity search. Filessuperimpose.sXX.list(e.g.superimpose.s40.list) correspond to a non-redundant set of protein structures selected at a certain threshold of pairwise sequence similarity (40% in the provided example). The selected proteins are marked by the '*' sign in the 'NR' (i.e., non-redundant) column;superimpose.fastaandsuperimpose.sXX.seq- files with sequences of all proteins selected during the structure similarity search, and sequences of proteins from the non-redundant sets selected at a certain threshold of pairwise sequence similarity, respectively;PDBID1__PDBID2.fasta- files with sequence representation of pairwise structural alignments;results_nr95/- folder with PDBs of proteins from the 95%-non-redundant set;results_nr95_ordered/- folder with PDBs of proteins from the 95%-non-redundant set renamed by adding a prefix which corresponds to the rank in thesuperimpose.listfile (i.e., reflecting similarity to the query);results_selected/- folder with PDBs of proteins from the representative set selected to create the core structural alignment;postgresql.log- database read/write log.
The seqsearch_TaskID.tar.gz archive has the following content:
PDBID.final.fasta- files with sequence alignments of proteins selected by sequence similarity to proteins PDBID from the core structural alignment. These files will be used at the Step 4 of the Protocol to build the final alignment;
If the use of the MAPU tool was enabled, thePDBID.final.fastafiles will contain the MAPU-processed (trimmed) versions of the sequence alignments. The original (full) sequence alignments will be present asPDBID.final.fasta_bakand the MAPU processing logs will be available asPDBID.final.mapu.log;BLAST_PDBID/- folders with the results of sequence similarity searches using proteins from the core structural alignment as a query (PDBID ).
EachBLAST_PDBID/folder contains the following files and folders:PDBID.seq- sequence of the representative protein from the core structural alignment, which was used as a query to run sequence similarity search;uniprot_sprot/anduniprot_trembl/- folders with the raw results of blastp of the query protein in the corresponding database:- The
uniprot_sprot/folder contains the query protein sequencePDBID.seq, theblosum45/,blosum62/, andblosum80/subfolders with the blastp output log files, andRESULTS.seqfile with collected sequences; - The
uniprot_trembl/folder contains the same files andblosum62/subfolder with blastp output log.
- The
RESULTS.seq- file with all protein sequences selected above the threshold of the blastp searchPDBID.blast.seq- file with sequences selected by the Sequence length filter;PDBID.blast.s95.seq- file with sequences selected by the Redundancy filter;PDBID.blast.sXX.fasta- file with aligned sequences selected by the Redundancy filter at the XX% similarity threshold;PDBID.blast.sXX.fasta- file with aligned sequences selected by the Redundancy filter at the XX% similarity threshold;PDBID.blast.sXX.hY.YY.seq- file with aligned sequences selected by the Dissimilarity filter at the Y.YY bit score per column threshold;PDBID.blast.sXX.hY.YY.fasta- file with aligned sequences selected by the Dissimilarity filter at the Y.YY bit score per column threshold;PDBID.blast.sXX.hY.YY.fasta- file with aligned sequences selected by the Dissimilarity filter at the Y.YY bit score per column threshold;seqsearch_PDBID.stdout.logandseqsearch_PDBID.stderr.log- Standard output and standard error output log files.
seqsearch_reassign.log- log of reassignment operations (if a protein was selected by multiple sequence similarity searches with different queries this file describes what copies of that protein were removed and which one was retained);seqsearch_parm.log- file with parameters used for each sequence similarity search.
Analysis section: Basic alignment statistics
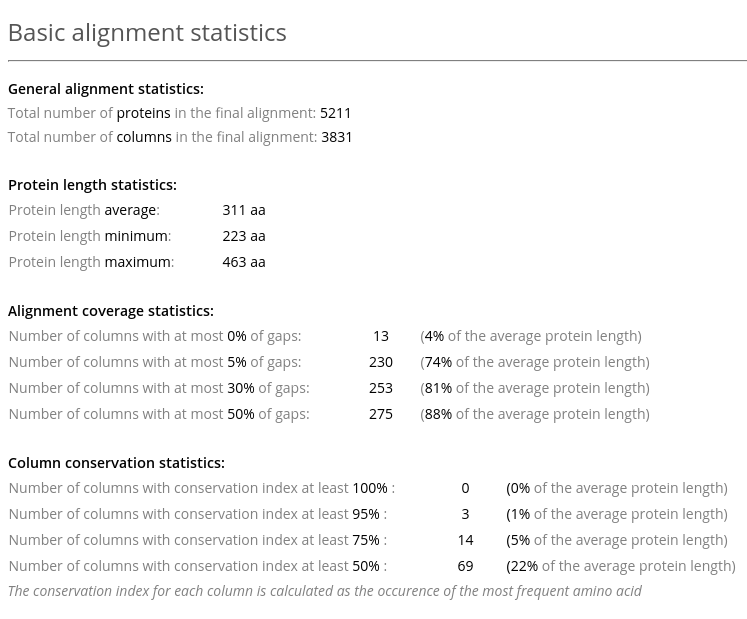
The Basic alignment statistics in Analysis section provides information about the number of proteins in the alignment, their length, and general information about the alignment quality (see an example above). The latter includes statistics on gaps and conserved residues.
Analysis section: Sequence analysis
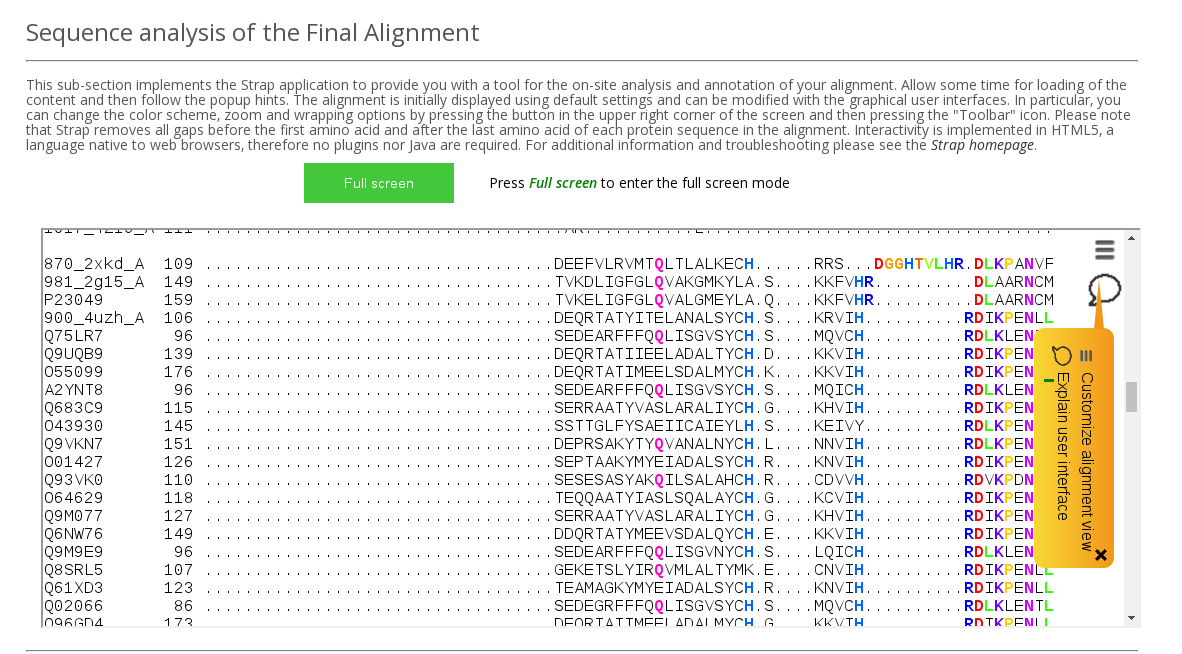
Sequence analysis of the Final Alignment field in the Analysis section implements Strap application to provide you with a tool for the on-site analysis and annotation of your alignment (see an example above). Allow some time for loading of the content and then follow the popup hints. The alignment is initially displayed using default settings and can be modified with the graphical user interface. In particular, you can change the color scheme, zoom and wrapping options by using the Toolbar functionality. Please note that Strap removes all gaps before the first amino acid and after the last amino acid of each protein sequence in the alignment. Interactivity is implemented in HTML5, a language native to web browsers, therefore no plugins nor Java are required.
Analysis section: Structure-based annotation

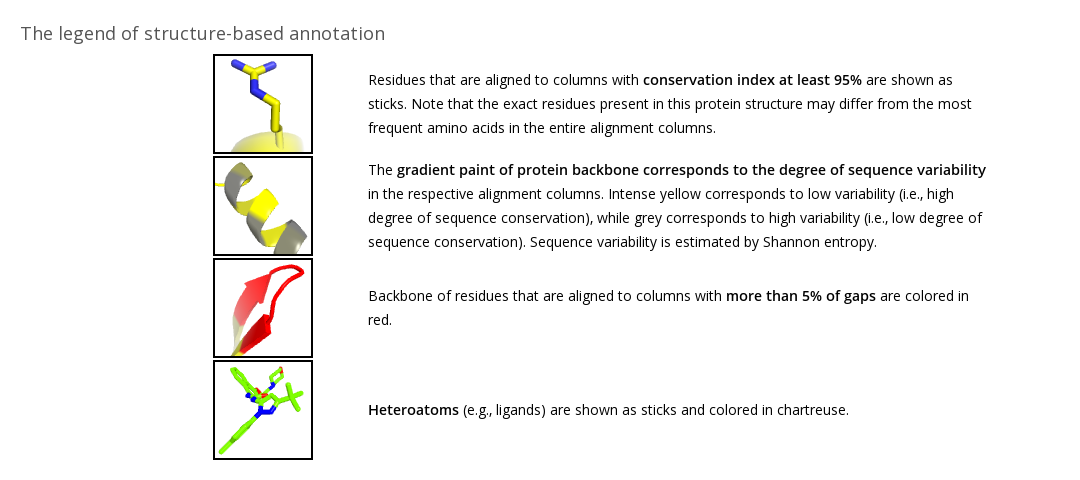
Structure-based annotation of the Final Alignment field (available in Mode 1 and Mode 4) in the Analysis section implements the JSMol application to provide you with a tool for the structure-based analysis of your alignment (see an example above). Each representative protein structure in the core structural alignment is annotated according to the final alignment (see the legend). You can choose a protein from the dropdown menu and allow some time for loading of the content. The structure-based annotation is intended to estimate the alignment quality as well as for preliminary study of your protein families by visualizing the basic alignment statistics on a particular protein structure. Amino acids of the selected protein that correspond to alignment columns with more than 5% of gaps are colored in red, indicating parts of the protein that may not have an equivalent in other homologs within a superfamily. Amino acids of the selected protein that correspond to alignment columns with at least 95% conservation index (i.e., the same amino acid is present in at least 95% of homologs) are colored in yellow and shown as sticks, indicating parts of the protein that are constant in most aligned homologs. The yellow-to-grey gradient paint corresponds to different conservation index from 95% to 50%, respectively. You can rotate the protein and zoom in for a closer look at the area of interest. Interactivity is implemented in HTML5, a language native to web browsers, therefore no plugins nor Java are required.
The Structure-based annotation of the Final Alignment is not available in Modes 2 and 3 because in these cases the Mustguseal task does not include any PDB files. For your convenience the annotation script can be downloaded for local use. This is the same script which is implemented to create the Structure-based annotation at the Mustguseal Analysis section. Instructions and examples are included in the archive.