mpiWrapper
Parallel workflow manager for non-parallel bioinformatic applications to solve large-scale biological problems on a supercomputer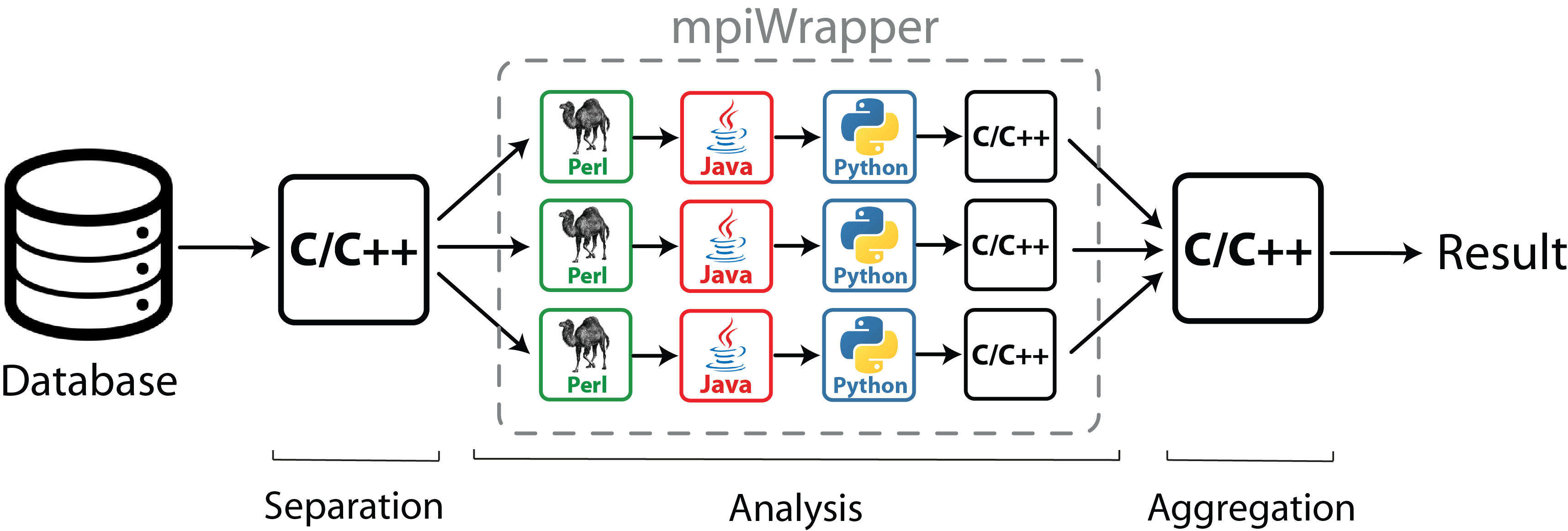
This software is intended to solve large computational problems which can be divided into independent subtasks to be analyzed separately without interprocess communication
THIS WEB-SITE/WEB-SERVER/SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS OR HARDWARE OWNERS OR WEB-SITE/WEB-SERVER/SOFTWARE MAINTEINERS/ADMINISTRATORS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE WEB-SITE/WEB-SERVER/SOFTWARE OR THE USE OR OTHER DEALINGS IN THE WEB-SITE/WEB-SERVER/SOFTWARE. PLEASE NOTE THAT OUR WEBSITE COLLECTS STANDARD APACHE2 LOGS, INCLUDING DATES, TIMES, IP ADDRESSES, AND SPECIFIC WEB ADDRESSES ACCESSED. THIS DATA IS STORED FOR APPROXIMATELY ONE YEAR FOR SECURITY AND ANALYTICAL PURPOSES. YOUR PRIVACY IS IMPORTANT TO US, AND WE USE THIS INFORMATION SOLELY FOR WEBSITE IMPROVEMENT AND PROTECTION AGAINST POTENTIAL THREATS. BY USING OUR SITE, YOU CONSENT TO THIS DATA COLLECTION.
Navigation:
- Description
- Publication
- Background
- Applicability
- Download
- Installation
- Usage
- Troubleshooting
- License
- Acknowledgements
The mpiWrapper is a software to manage execution of nonparallel programs in the parallel mode – on a Desktop station powered by a multi-core CPU or a supercomputer running Linux/Unix-based operating system. The software is intended to solve large computational problems which can be divided into independent subtasks to be analyzed separately without interprocess communication. The mpiWrapper has to be launched as a regular MPI program by the appropriate MPI utility on a local host or a supercomputer. The allocated resources are then used by the mpiWrapper to manage parallel execution of the provided singleCPU subtasks. The program can be implemented to launch all conventional "single-CPU" Unix/Linux applications, including scientific programs as well as Perl, Python, and shell scripts. No rewriting of the original source code is necessary to execute an application with the mpiWrapper. The mpiWrapper is easy to use, does not require special installation procedures and supports re-submission of subtasks on node failure. Computational efficiency of the mpiWrapper on a large number of CPUs is approaching its maximum value as a result of no interprocess communication/synchronization between individual subtasks during their execution, and minimal overhead for task management.
D. Suplatov, N. Popova, S. Zhumatiy, V. Voevodin, V. Švedas (2016) Parallel workflow manager for non-parallel bioinformatic applications to solve large-scale biological problems on a supercomputer, J Bioinform Comput Biol, 14(2), 1641008, doi:10.1142/S0219720016410080.
In this publication we discuss the hybrid approach in both the hardware and software setup as the probable strategy to solve large-scale biological problems taking into account the diversity of applications and algorithms in computational life sciences. From the software perspective, an efficient solution to analysis of increasing amounts of biological data requires both a wise parallel implementation of resource-hungry algorithms as well as a smart layout to manage multiple invocations of relatively fast algorithms. In this context we introduce the algorithm of the mpiWrapper - a software to manage efficient execution of non-parallel implementations of scientific algorithms in a parallel setup. We discuss a wide range of scientific applications of the program and explain its potential in efficiently processing huge amounts of biological data on a supercomputer. In addition, we report a case-study of substrate specificity of an enzyme using the mpiWrapper to run the task in parallel. This case-study is important to show the potential of the program to accelerate analysis of real biological data.
Rapid expansion of online resources providing access to genomic, structural and functional information associated with biological macromolecules opens an opportunity for a deeper understanding of the mechanisms of biological processes by systematically analyzing large amounts of data, but requires novel strategies to optimally utilize computer processing power. Various bioinformatic and molecular modeling methods have been developed to date. Some algorithms require extensive computational resources - because they deal with the complete dataset in order to divide it into subtasks or merge the output from individual subtasks ("Separation" and "Aggregation", see the scheme). However a substantial part of bioinformatic and molecular modeling approaches have fast implementations which take at most several hours to analyze a common input on a modern desktop station. These are organized into pipelines of programs and scripts (usually written in different programming languages) to analyze independent subtasks ("Analysis", see the scheme). Each subtask can run fast even on a single CPU, however due to multiple invocations for a large number of subtasks the whole setup requires a significant computing power.
The mpiWrapper has a wide range of scientific applications. It can be used to launch all conventional Unix/Linux programs without the need to modify their original source codes. E.g., the mpiWrapper can be used to execute molecular docking of millions of ligands using non-parallel build of Autodock, manage the prediction of a large library of protein mutants with Modeller, or even run multiple BLAST searches given that the nodes are equipped with the appropriate amount of memory. The mpiWrapper can be used to run repetitive jobs which are carried out to collect more data for statistical analysis.
Download C++ source code for mpiWrapper v.2.1.2 compatible with openmpi v. 2.1.1 and v. 3.0.0 (2017-12-10) [download]
Download C++ source code for mpiWrapper v.2.1.1 (2016-01-13) [download]
The mpiWrapper does not require any special installation procedures. Simply compile the code using the command and use the binary straight ahead:
mpicxx -pthread mpiwrapper.cpp -o mpiWrapperThe mpiWrapper was developed to manage multiple invocations of "single-CPU" programs. These programs can be written in different programming languages, organized into sequential pipelines and are expected to be relatively fast (up to a couple of days on a single core). Lets imagine that you want to screen a large in silico library of potential drugs using molecular docking. Or you want to annotate a genome by executing a sequence of fast bioinformatic routines on each gene. In both cases a single subtask (docking of one ligand library or analysis of one gene CDF data) can be quickly processed even on a single CPU. However, due to multiple invocations (for millions of ligands or millions of genes per genome) the whole setup requires extensive computational resources. This is when the mpiWrapper can help by encapsulating these individual threads (see the scheme above) and launching them in a parallel mode on a supercomputer or a fast multi-core Desktop CPU.
General usage
The command to start mpiWrapper is:
mpilauncher [options] /path/to/mpiWrapper /path/to/taskfile.txt
where mpilauncher depends on you particular system and its [options] have to specify the number of CPUs (CPU cores) to be used by the mpiWrapper to process all subtasks given in the taskfile.txt.
Basic examples
- Launch mpiWrapper locally on 8 CPU cores:
mpirun -hosts localhost -np 8 mpiWrapper taskfile.txt - Launch mpiWrapper compiled with IntelMPI on 64 CPU cores of a supercomputer with sbatch and impi batch script:
sbatch -np 64 impi mpiWrapper taskfile.txt - Launch mpiWrapper on 64 CPU cores of a supercomputer using cleo queue manager:
cleo-submit -np 64 mpiWrapper taskfile.txt - Note: mpirun, sbatch and cleo-submit are not part of the mpiWrapper. These commands are widely used to launch MPI applications in parallel and are included in distributions of the corresponding software packages. E.g., you can try a free openMPI software package - an open source implementation of the Message Passing Interface.
Preparation of the task file
The task file is a plain text file that contains subtasks - commands to be executed one per line. Each line of the task file will be treated by the mpiWrapper as an individual subtask. Each line of the task file starts with a full path to the executable (a binary or a script which starts from '#! path/to/interpreter') followed by a list of flags/command line parameters. Multiple commands separated by ';' as well as I/O redirections using the '|' are not allowed in the taskfile - these should be encapsulated into a bash script and then path to this script should be provided in the taskfile. Each subtask will be sent for processing to a separate CPU.
An example of the task file:
/path/to/executable_1.bin -i [some_input_1] -o [output_1]
/path/to/executable_1.bin -i [some_input_2] -o [output_2]
/path/to/executable_2.bin -i [some_input_3] -o [output_3]
/path/to/executable_2.bin -i [some_input_4] -o [output_4]
Note: Each subtask is executed on a single CPU. Therefore it seems logical that the number of subtasks in the task file has to be equal or larger than the number of requested CPUs. Submitting less subtasks than CPUs will obviously lead to non-productive use of computer power (extra CPUs will be blocked but not used). In this case the mpiWrapper will throw a warning but continue.
Managing the standard output
It is expected that every subtask will produce an output file which will be printed to the hard drive. In addition, every program generates a standard output stream to share information with the user - e.g. the program title, version, current progress, etc. In most cases such output would be of no practical value. Nevertheless, this second output stream would put additional pressure on the resources if used extensively, namely the file system and the communicator. Therefore, unless the standard output of a subtask is known to contain important information, it is advised to minimize or nullify this output stream. There are two ways to do so. First option would be to use an appropriate flag to switch a program into the quiet mode (e.g. -q or -quiet). Obviously, this option is subjected to availability and not every program has the appropriate flag. So, the second option is to wrap your subtasks inside a script which re-routes the output to /dev/null.
#!/bin/bash
input=$1
output=$2
binary="/path/to/program1"
$binary -i $input -o $output >& /dev/null
To execute multiple invocations of a sequence of independent programs, when the output of one program is the input to the next program, using mpiWrapper the users are encouraged to encapsulate such pipelines into a shell script. Execution of these shell scripts should be further specified as subtasks in the taskfile for the mpiWrapper. A pseudocode example of such bash script is provided below.
#!/bin/bash
input=$1
output=$2
binary1="/path/to/program1"
binary2="/path/to/program2"
binary3="/path/to/program3"
#Launch the first program
$binary1 -i $input -o temp_output_1
#Launch the second program
$binary2 -i temp_output_1 -o temp_output_2
#Launch the final program
$binary3 -i temp_output_2 -o $output
There are reasons for a command to fail when executed on a CPU, e.g. memory error or problems with the shared file system on the particular node, etc. By default, the mpiWrapper considers a non-zero exit code as a failure. In this case the subtask is returned to the queue to be resubmitted on a different CPU. A CPU that failed to execute 5 subtasks will be blocked for 60 minutes. However, a non-zero exit status does not necessarily mean that something is wrong with the CPU. It may simply occur as a result of incorrect command syntax or lack of the specified input files, etc. Therefore, by default, a subtask that failed 10 time is removed from the queue and flagged as failed in the final statistics printed by the mpiWrapper. To customize the "re-submission on failure" function of the mpiWrapper users are encouraged to launch their subtasks using a dedicated bash script that will control the cases when a non-zero exit status is reported to the mpiWrapper. In other words, this script has to distinguish situations when subtask failure occurs because of a possible CPU malfunction (then re-submission of a subtask on a different "healthy" node can help) from hardware unrelated cases (e.g., absence of input files or wrong syntax). A pseudocode example of such bash script is provided below.
#!/bin/bash
binary=$1
input=$2
output=$3
#If the binary does not exist or is not executable it is useless to resubmit the subtask
if ($binary !-exists OR !-is_executable) {exit 0;}
#If the input is not accessible it is useless to resubmit the subtask
if ($input !-exists OR !-is_readable) {exit 0;}
#If the output file is not writable it is useless to resubmit the subtask
if ($output !-writable) {exit 0;}
#Execute the binary with parameters
$binary -i $input -o $output
#Return exit code to the mpiWrapper
#If this is a non-zero exit code it might be an internal hardware problem of this particular node
#Then the mpiWrapper will re-submit this subtask on a different node
exit $?
Problem: You execute the mpiWrapper on your multi-core desktop but it yet runs on just one core
Problem: You do not understand how to provide the number of CPUs to run on to the mpiWrapper
Solution: You might have executed the mpiWrapper like this:
Problem: You execute the mpiWrapper but it reports that all task have failed
Solution: Each like of the taskfile must start with a full path to the executable (a binary or a script which starts from '#! path/to/interpreter') followed by a list of flags/command line parameters. Multiple commands separated by ';' as well as I/O redirections using the '|' are not allowed in the taskfile - these should be encapsulated into a bash script and then path to this script should be provided in the taskfile. Therefore, there could be two likely explanations for the above-mentioned problem. First, you used the wrong syntax to write you commands. E.g., you specified the wrong path to the binary, or the binary is not 'executable' (use chmod), or you messed up the flags, etc. The second explanation is that mpiWrapper requires the full path to the executable to be specified in the taskfile, and you might have used the local one.
This program is a free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or of any later version.
This work was supported by the Russian Foundation for Basic Research [grant #14-07-00437] and the Russian Science Foundation [grant #15-14-00069]