visualCMAT
A web-server to select and interpret
correlated mutations/co-evolving residues in protein families
Have a protein family? ...

... to predict and visualize
correlated mutations/co-evolving residues
in protein structures
Version 1.0; August 4th 2017
THIS WEB-SITE/WEB-SERVER/SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS OR HARDWARE OWNERS OR WEB-SITE/WEB-SERVER/SOFTWARE MAINTEINERS/ADMINISTRATORS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE WEB-SITE/WEB-SERVER/SOFTWARE OR THE USE OR OTHER DEALINGS IN THE WEB-SITE/WEB-SERVER/SOFTWARE. PLEASE NOTE THAT OUR WEBSITE COLLECTS STANDARD APACHE2 LOGS, INCLUDING DATES, TIMES, IP ADDRESSES, AND SPECIFIC WEB ADDRESSES ACCESSED. THIS DATA IS STORED FOR APPROXIMATELY ONE YEAR FOR SECURITY AND ANALYTICAL PURPOSES. YOUR PRIVACY IS IMPORTANT TO US, AND WE USE THIS INFORMATION SOLELY FOR WEBSITE IMPROVEMENT AND PROTECTION AGAINST POTENTIAL THREATS. BY USING OUR SITE, YOU CONSENT TO THIS DATA COLLECTION.
Publication: Suplatov D., Sharapova Y., Timonina D., Kopylov K., Švedas V. (2018) The visualCMAT: a web-server to select and interpret correlated mutations/co-evolving residues in protein families. J Bioinform Comput Biol., 16(2), 1840005. DOI: 10.1142/S021972001840005X
Navigation:
- Prerequisites for using visualCMAT
- Preparing the input data
- The visualCMAT parameters
- The visualCMAT output
- Guidelines on working with the visualCMAT output
- The visualCMAT example
- The visualCMAT file sharing and security features
- Implementation of visualCMAT in the laboratory practice
- Citing visualCMAT
Highlights:
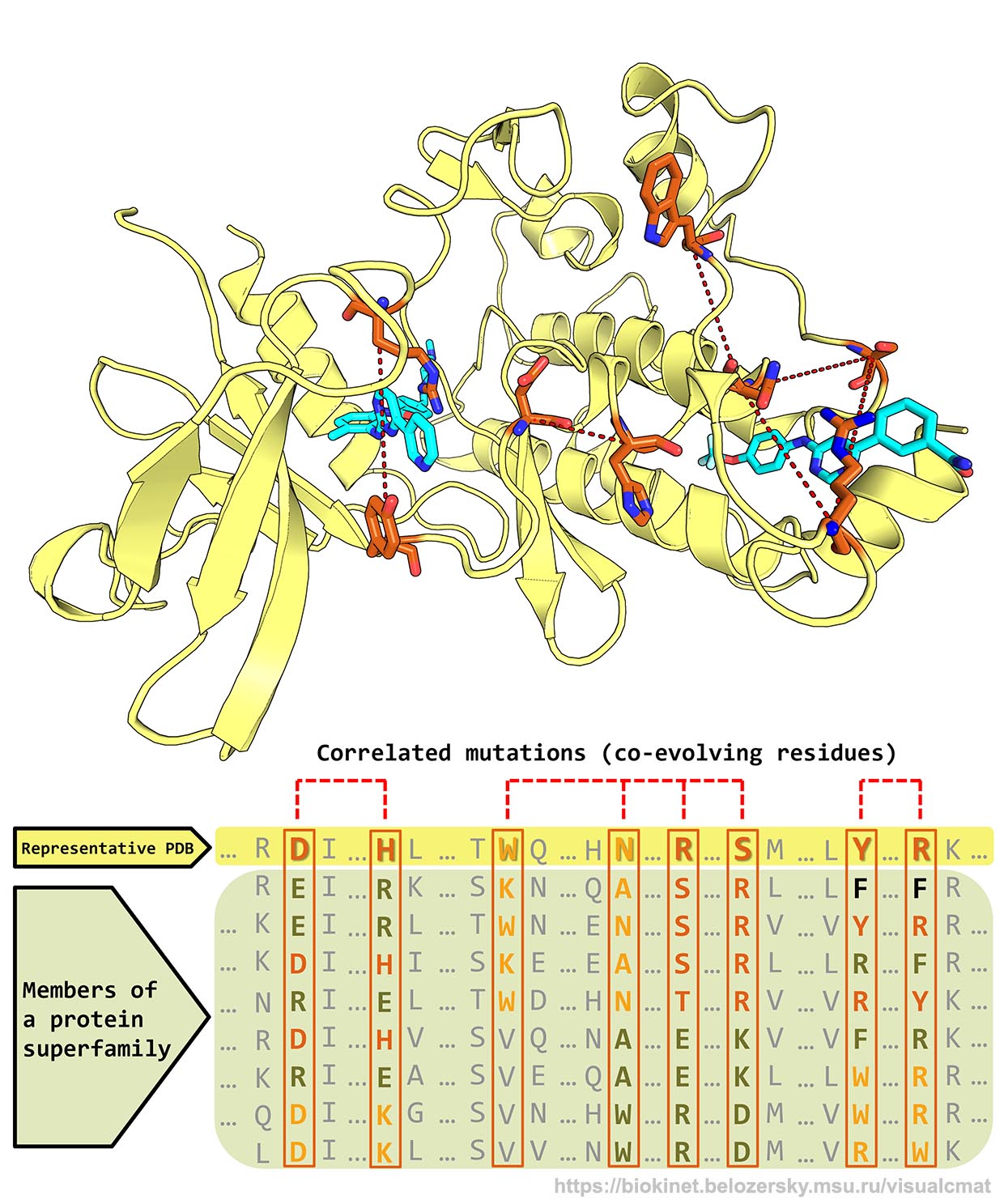
- The visualCMAT web-server was designed to assist experimental research in the fields of protein/enzyme biochemistry, protein engineering, and drug discovery by providing an intuitive and easy-to-use interface to the correlated mutations analysis;
- The visualCMAT web-server can be used to understand the relationship between structure and function and identify co-evolution patterns in protein superfamilies, implemented at selecting hotspots and compensatory mutations for rational design and directed evolution experiments to produce novel enzymes with improved properties, and employed at studying the mechanism of selective ligand's binding and allosteric communication between topologically independent sites in protein structures;
- The input multiple alignment of a diverse set of related proteins can be automatically constructed by the sister web-server Mustguseal using all available information about their structures and sequences in public databases, and then directly submitted to the visualCMAT web-server in one click. Large structure-guided sequence alignments of functionally diverse families that include thousands of proteins can be automatically constructed using that public web-server;
- Press
 to run the Mustguseal web-server with a pre-selected set of parameters and automatically construct a multiple alignment of your protein families for further analysis by the visualCMAT;
to run the Mustguseal web-server with a pre-selected set of parameters and automatically construct a multiple alignment of your protein families for further analysis by the visualCMAT;
- Alternatively, you can manually submit a multiple protein alignment in FASTA format and a representative protein structure in PDB format to the visualCMAT server. Multiple alignments of thousands of proteins can be handled by this server;
- The visualCMAT server will automatically predict correlated substitutions in protein structures, classify them into physically interacting residues and long-range correlations, annotate and rank binding sites on the protein surface by the presence of statistically significant co-evolving positions;
- All steps of the visualCMAT server protocol are executed entirely on the server side using a combination of bioinformatic methods for sequence and structure analysis. You do not need any specific software on your side;
- The results of the visualCMAT are convenient for visual expert analysis and can be downloaded to a local computer as a single all-in-one PyMol session file with multiple layers of annotation corresponding to bioinformatic, statistical and structural analyzes of the predicted co-evolution, or operated on-line using the built-in interactive analysis tools. Interactivity is implemented in HTML5 and therefore no plugins nor Java are required;
- Press the Run visualCMAT on-line button and then press the Demo mode button to request a demonstration of the visualCMAT server.