Zebra examples
The Zebra was recognized as a computational tool to design and engineer biocatalysts [Chemical Communications, 2017, 53(2), 284-297, link; Curr. Opin. Chem. Biol., 2014, 19, 8–16, link] and used to assist the selection of hot-spots for protein engineering to introduce amidase activity into Candida antarctica Lipase B (CALB) [Protein Eng. Des. Sel., 2012, 25(11), 689–697, link], increase alkaline stability in Penicillin Acylase from Escherichia coli [PloS ONE, 2014, 9(6), e100643, link], improve the rate of asymmetric hydration of aliphatic alkenes by Oleate Hydratase from Elizabethkingia meningoseptica [Angewandte Chemie, 2019, 58(1), 173-177, link]; to select particular amino acids responsible for binding regulatory ligands and assist the design of novel inhibitors of the Arachidonic Acid Cascade [Molecular informatics, 2018, 37(3), 1700073, link]; to study the structure-function relationship in various protein superfamilies: [PloS one, 2017, 12(5), e0177392, link], [J. Microbiology, 2018, 56, 246–254, link], [FEBS open bio, 2018, 8(6), 1013-1028, link], etc. Here we provide the guidelines and data to quickly reproduce two case-studies of protein superfamilies using Zebra.
Examples:
- Example 1 (small alignment): Superfamily of Glutathione S-transferases
- Example 2 (large alignment): Superfamily of the fold type I PLP‐dependent enzymes
Example 1 (small alignment): Superfamily of Glutathione S-transferases
This example features a small alignment of ~100 proteins to quickly test-run the Zebra pipeline
Background. Glutathione S-transferases (GST) represent a superfamily of detoxifying dimeric enzymes that are found in a wide range of organisms and catalyze the transfer of tripeptide glutathione to a variety of electrophilic co-substrates. Each subunit of a protein dimer consists of two structurally distinct domains and contains the independent catalytic center composed of the G-site for binding structurally conserved physiological substrate glutathione and the H-site for binding structurally diverse endogenous and xenobiotic substances.
Collection and alignment of homologs. GSTs that belong to nine classes Alpha, Mu, Pi, Sigma, Phi, Tau, Theta, Zeta, and Omega were collected and aligned as previously discussed [J. Biomol. Struct. Dyn., 2014, 32(11), 1752-1758, link]
Input data to Zebra web-server. Download the multiple sequence alignment and the representative protein PDB structure using the links below, upload them to Zebra in the "Mode 1", and press "Submit":
- Download the alignment containing 103 sequences and structures of proteins belonging to the nine classes of GSTs: [download]
- Download the PDB structure 2GST of the Rat Mu class GST: [download]
Running time. The running time of this task is ~30 seconds
Output. The hallmark of Zebra2 is the interactive analysis toolkit. To operate this example on-line go to the Zebra2 submission page and press the Demo mode (GST) button. The results can also be downloaded to a local computer for an off-line analysis:
- Download the text version of the Zebra results: [download]
- Download the Zebra 3D-annotation files: [download]
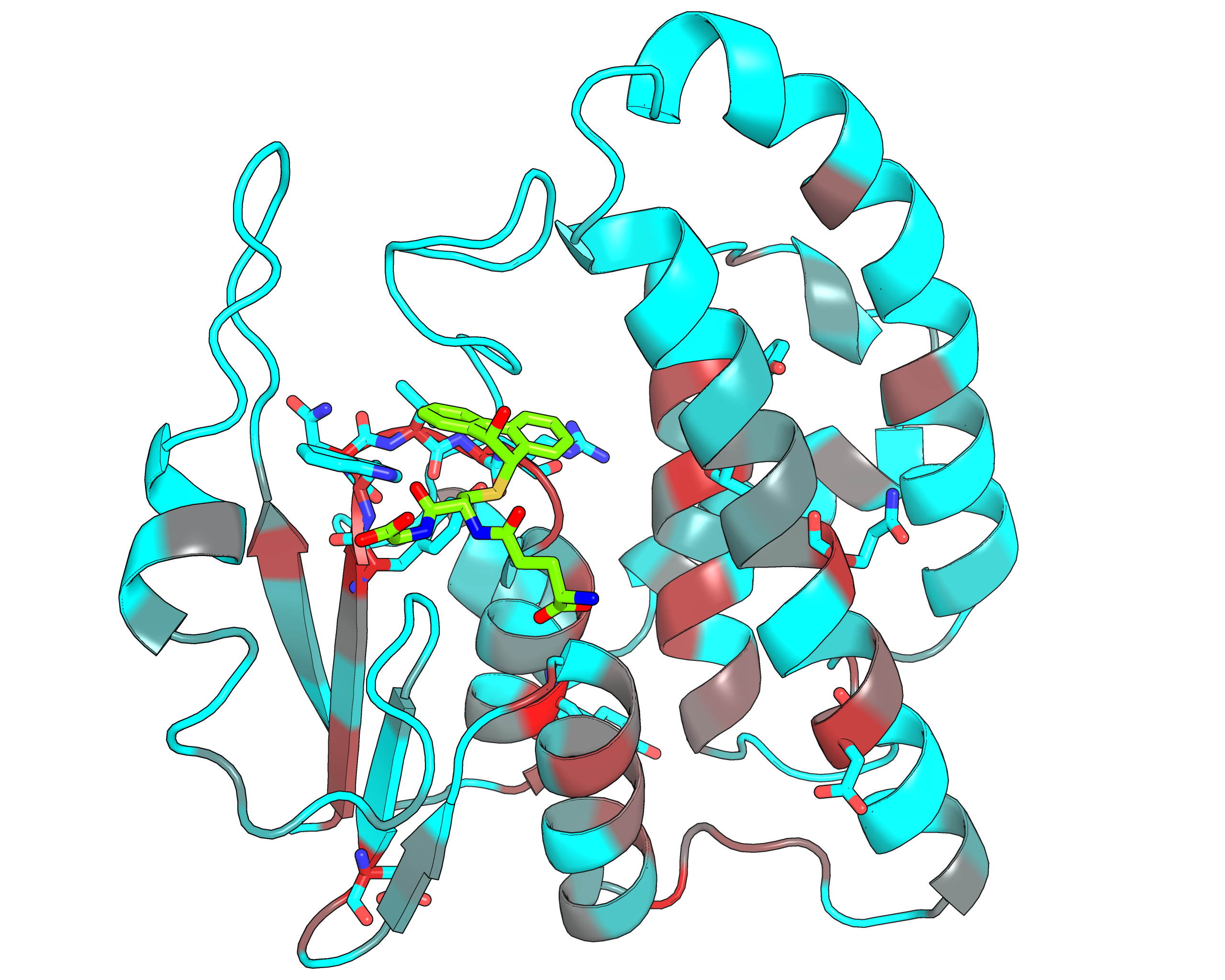 |
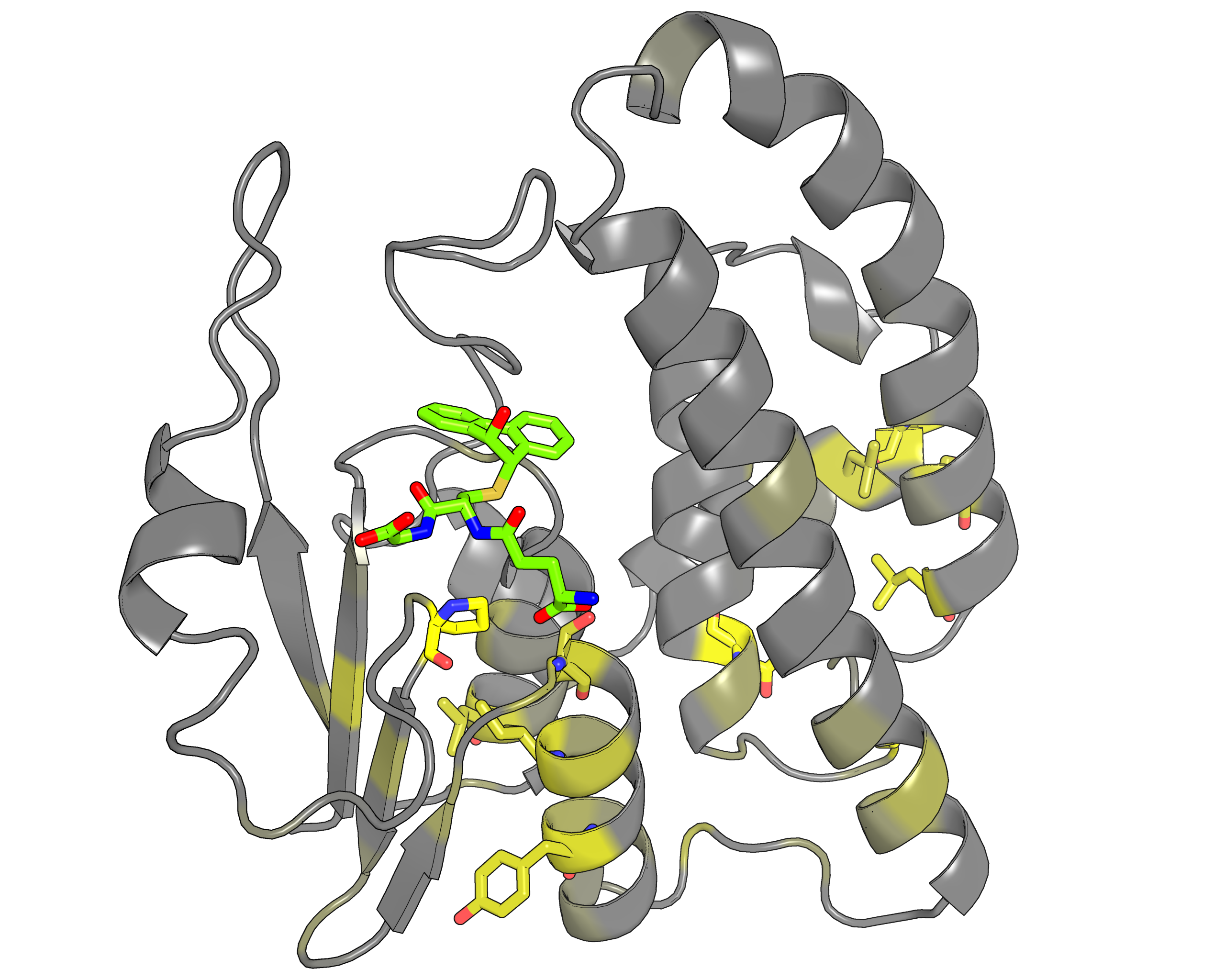 |
| Representative protein 3D-structure gradient-painted according to specificity of positions among different GST classes | Representative protein 3D-structure gradient-painted according to conservation of positions among different GST classes |
Results and interpretation. Two subfamily classifications were automatically predicted by the server that correspond to previously discussed functional classification of GST enzymes [Genome biology, 3(3), reviews3004-1, link]. First classification features animal-specific Alpha/Mu/Pi/Sigma classes united into one specificity group, and mammalian Omega, plant-specific Tau/Phi and mixed Theta/Zeta classes defined as separate groups. The second classification features nine subfamilies for each GST class. Based on the bioinformatic analysis it was suggested that the SSPs moderate functional diversity in the GST superfamily in two ways – by providing a specific interaction pattern with a substrate through sequence variability of the H-site, and through different conformational plasticity of the active site mediated by surrounding structural environment. The details of this example were published in a separate paper [J. Biomol. Struct. Dyn., 2014, 32(11), 1752-1758, link].
Example 2 (large alignment): Superfamily of the fold type I PLP‐dependent enzymes
This example features a large alignment of ~4185 proteins that is qualitatively similar to was has been recently used to reveal determinants of reaction specificity and design promiscuous catalytic activity in the fold-type I PLP-dependent enzymes [FEBS open bio, 2018, 8(6), 1013-1028, link]
Background. PLP‐dependent enzymes are mainly involved in the amino acid metabolism in all living organisms and catalyse a number of diverse chemical reactions, such as decarboxylation, transamination, racemization, β‐elimination, carbon‐carbon bond cleavage and formation. Due to their catalytic versatility, the PLP‐enzymes have been exploited as biocatalysts for the production of natural and non‐natural amino acids and amino acid derivatives. The aspartate aminotransferase superfamily is the largest group of PLP‐dependent enzymes which implement versatile catalytic activities (aldolase, transaminase, decarboxylase, etc.) within a common structural framework commonly being referred to as the Fold type I. Until recently, the amino acid residues which play a key role in determining the reaction specificity in the aspartate aminotransferase superfamily were poorly characterized.
Collection and alignment of homologs. This step was performed automatically using the Mustguseal web-server. The PDB structure 3WGB (chain A) of L‐threonine aldolase from Aeromonas jandaei was submitted to the Mustguseal web-server in "Mode 1" and "Scenario 2" (see Zebra Input for details) to automatically construct an alignment of a non-redundant set of 4185 proteins with high structural but low sequence similarity to the query. This multiple alignment and the PDB structure 3WGB of the query protein were further submitted to the Zebra web-server for bioinformatic analysis.
Input data to Zebra web-server. Download the multiple sequence alignment and the representative protein PDB structure using the links below, upload them to Zebra in the "Mode 1", set the Min size of a subfamily (%) parameter to 4 (i.e., 4%) - see the explanation here, and press "Submit":
- Download the alignment containing 4185 sequences and structures of the fold-type I PLP-dependent enzymes with high structural but low sequence similarity to L‐threonine aldolase from Aeromonas jandaei: [download]
- Download the PDB structure of L‐threonine aldolase from Aeromonas jandaei: [download]
Running time. The running time of this task is ~30 minutes
Output. The hallmark of Zebra2 is the interactive analysis toolkit. To operate this example on-line go to the Zebra2 submission page and press the Demo mode (PLP) button. The results can also be downloaded to a local computer for an off-line analysis:
- Download the text version of the Zebra results: [download]
- Download the Zebra 3D-annotation files: [download]
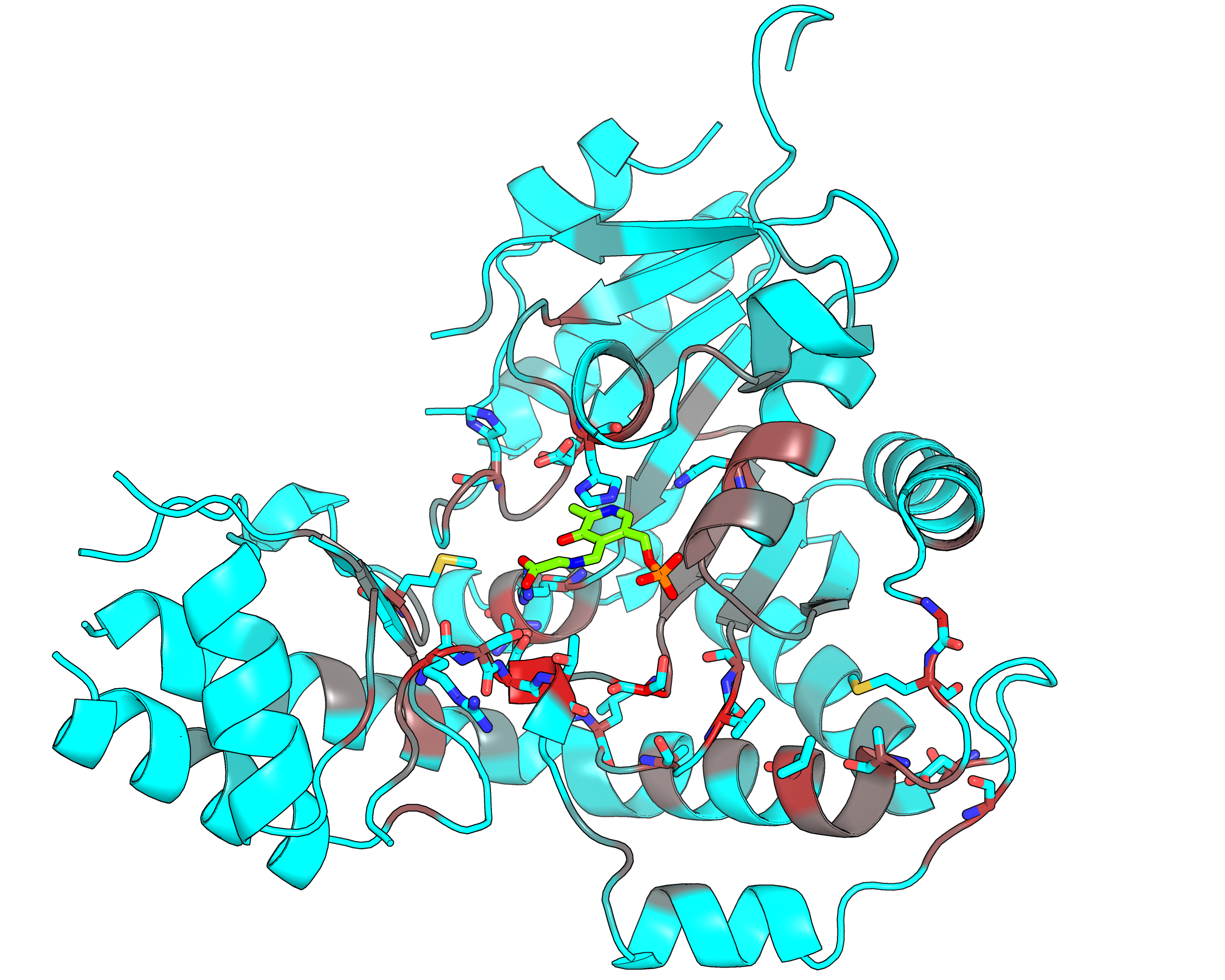 |
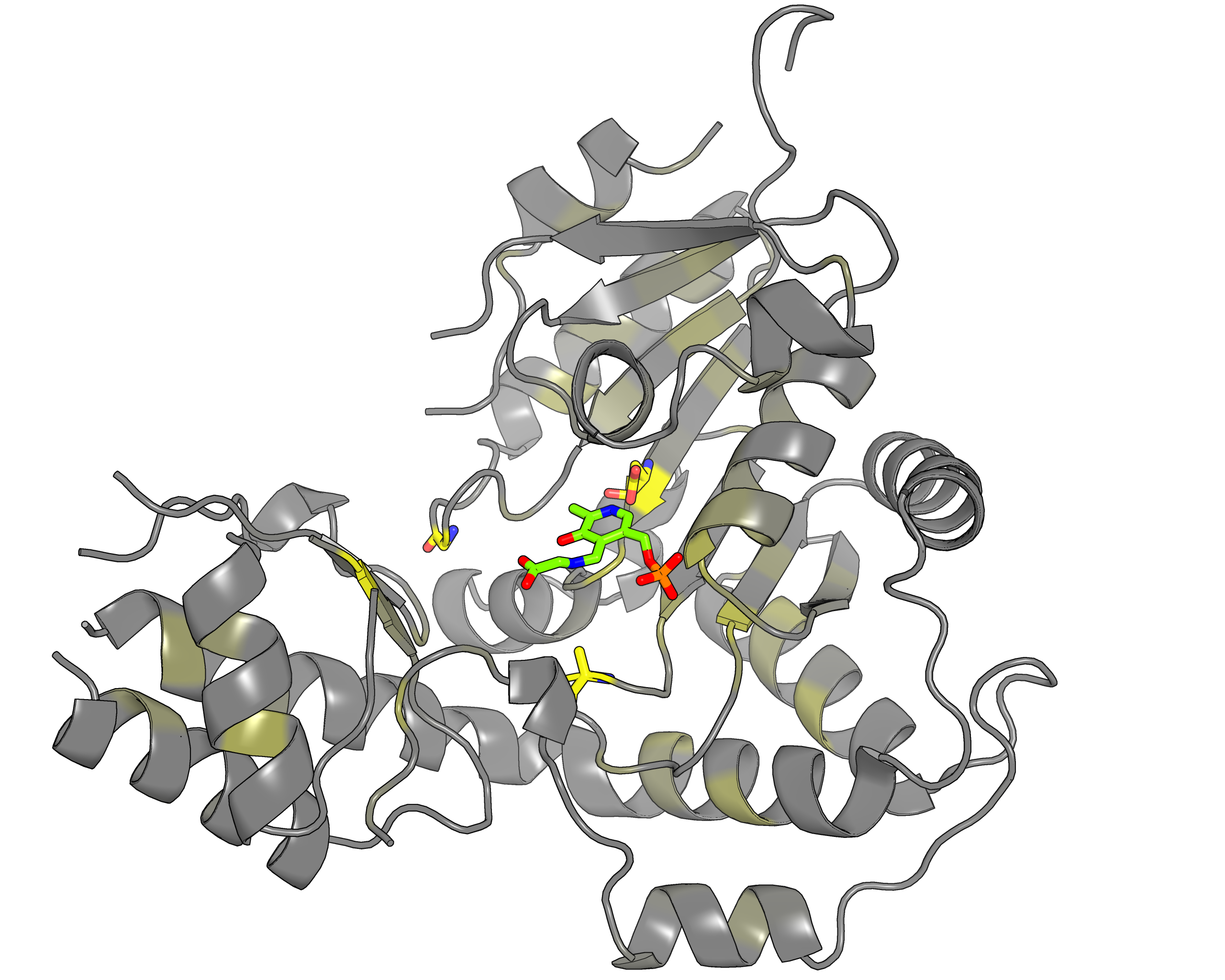 |
| Representative protein 3D-structure gradient-painted according to specificity of positions among PLP families with different reaction specificity | Representative protein 3D-structure gradient-painted according to conservation of positions among PLP families with different reaction specificity |
Results and interpretation. Mustguseal and Zebra were used to collect and study a large representative set of the aspartate aminotransferase superfamily with high structural, but low sequence similarity to L‐threonine aldolase from Aeromonas jandaei (LTAaj), in order to identify conserved positions that provide general properties in the superfamily, and to reveal family‐specific positions (FSPs) responsible for functional diversity. The roles of the identified residues in the catalytic mechanism and reaction specificity of LTAaj were then studied by experimental site‐directed mutagenesis and molecular modelling. It was shown that FSPs determine reaction specificity by coordinating the PLP cofactor in the enzyme's active centre, thus influencing its activation and the tautomeric equilibrium of the intermediates, which can be used as hotspots to modulate the protein's functional properties. Mutagenesis at the selected FSPs in LTAaj led to a reduction in a native catalytic activity and increased the rate of promiscuous reactions. The results provide insight into the structural basis of catalytic promiscuity of the PLP‐dependent enzymes and demonstrate the potential of bioinformatic analysis in studying structure–function relationship in protein superfamilies. See our recent publication for details [FEBS open bio, 2018, 8(6), 1013-1028, link].