Zebra parameters
Zebra parameters can be classified into three sections: (1) select positions (columns in the multiple alignment) that will be used to suggest classifications of proteins into functional subfamilies and to estimate the statistical significance of the corresponding subfamily-specific positions; (2) define parameters to improve the scoring and statistical assessment of the subfamily-specific positions; and, finally, (3) setup the algorithm to automatically classify proteins into functional subfamilies, or provide the manual classification file.
- Select positions for further bioinformatic analysis
- Set 3D-mode and number of random shuffles
- Set algorithm to classify proteins into functional subfamilies
Select positions for further bioinformatic analysis

Select positions (columns in the multiple alignment) that will be used to suggest classifications of proteins into functional subfamilies and to estimate the statistical significance of the corresponding subfamily-specific and conserved positions. First, define the allowed gap content. By default, all columns with more then 5% of gaps are dismissed from further consideration (i.e., they will be classified as "SKIPPED" and colored in white in the 3D-annotations of the results). Generally speaking, columns overpopulated by gaps are typically dismissed by the bioinformatic software for protein alignment analysis as low-informative and due to limitations of statistical models that assess the significance of amino acid substitutions in homologs. You should not set this threshold to a high value (i.e., to allow columns with a high content of gaps), unless there is a reason.
Second, define the columns to be considered for the bioinformatic analysis of the subfamily-specific positions. This selection will not affect the calculation of the conserved positions (i.e., all columns in the multiple alignment that pass the gap threshold will be assessed for conservation) and the sequence similarity network (i.e., SSN is calculated based on global pairwise sequence identities). This selection will affect the calculation of the subfamily-specific positions in three ways: (1) the automatic classification of proteins into subfamilies by the Zebra internal algorithm will be based on the sequence similarity between proteins only within the selected alignment columns, (2) the calculation of specificity Z-scores will be performed only for the selected alignment columns, leading to (3) the correction of the statistical model for subfamily-specific positions being limited only to the selected alignment columns. The default option is to "Consider all columns" of the multiple alignment that pass the gap filter. Alternatively, you can limit the columns to be considered for a particular purpose. E.g., select only the active site residues that participate only in binding of a particular moiety of a substrate to predict functional subfamilies and specific positions that define binding specificity only to the respective moiety.
You can make a selection of columns in two ways: by selecting all residues in close proximity to a crystallographic ligand (e.g., a substrate or water molecule), or by defining them explicitly via column IDs. Use the "Select columns by residues around a ligand in PDB" option, enter the name and residue ID of the ligand and specify the radius to select the amino acid residues around this ligand (i.e., assuming that the user-uploaded PDB file contains the respective ligand):
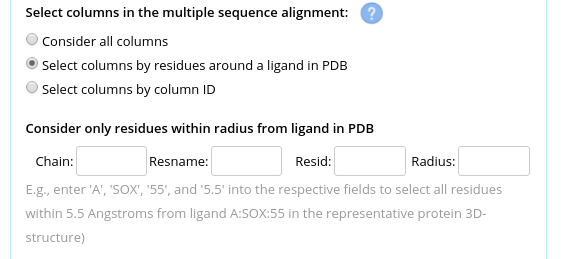
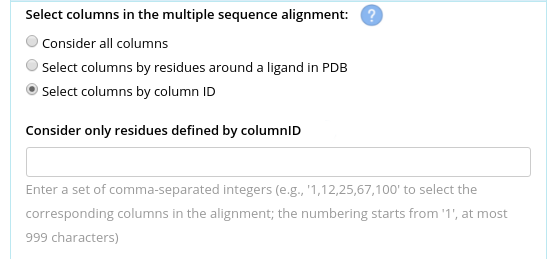
Another options is to explicitly provide a list of column IDs to be considered. The columns IDs start from 1 and correspond to their actual appearance in the multiple alignment file. Use the "Select columns by column ID" option and provide a string of comma-separated integers with no spaces, e.g.: "1,12,25,67,100".
Set 3D-mode and number of random shuffles
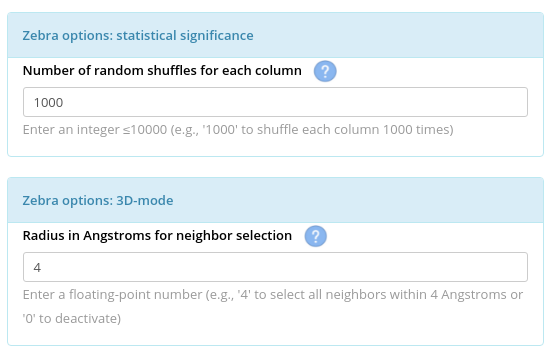
Each selected column of the multiple alignment will be independently shuffled N times to calculate the background statistical model. Then, the specificity/conservation scores of the real columns will be compared with the shuffled random models to calculate Z-scores and rank the conserved/subfamily-specific positions by statistical significance. The default is to perform 1000 shuffles per column.
3D-mode. When a three-dimensional structure of one of the family members is available (i.e., the "Mode 1"), Z-scores are further corrected to overvalue subfamily-specific positions that assemble into clusters with other subfamily-specific and conserved positions. The Z-score of the subfamily-specific position i is corrected by incorporating Z-scores of all conserved and subfamily-specific positions within the specified radius (the default is 4 Å). Benchmarking previously showed that the 3D-mode significantly improved the prediction accuracy of Zebra. The details are provided in the original Zebra publication.
Set algorithm to classify proteins into functional subfamilies
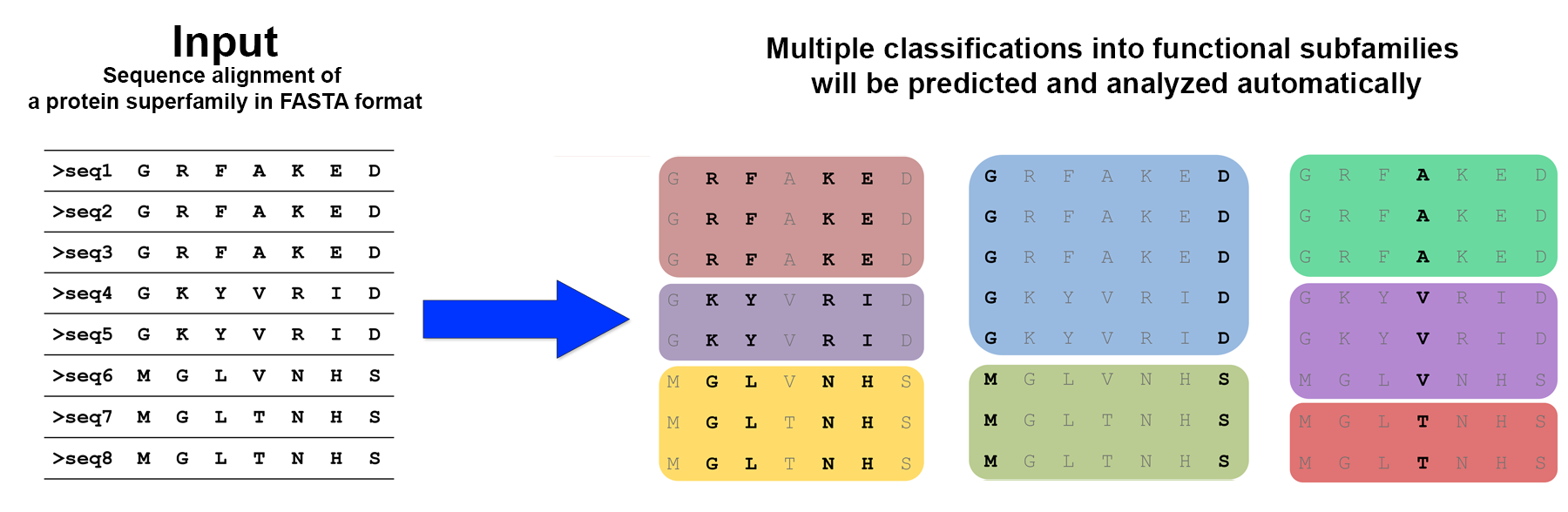
The calculate the subfamily-specific positions the input proteins have to be classified (i.e., divided) into subfamilies with different properties. The advantage of Zebra is that the algorithm can propose multiple classifications automatically by graph based clustering at different fragmentation levels, implement each classification to calculate the specific positions, and then summarize the results by ranking the automatically proposed subfamily classifications by the statistical significance of the specific positions they produce. This internal algorithm is used by default in the Zebra2 web-server.
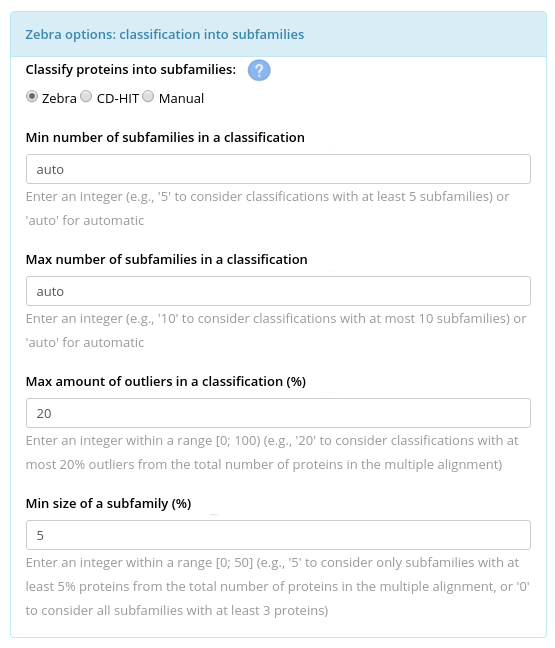
You can provide the following parameters to the Zebra internal classification algorithm: the expected minimum and maximum number of subfamilies in your input set of proteins (not limited by default); maximum amount of outliers per classification, i.e., the number of proteins that can be dismissed by the algorithm in attempt to produce a "better" classification (by default, 20%); and, finally, the minimum size of a subfamily (by default, 5%). The latter parameter means that if a specificity group will be automatically proposed during the automatic classification, but its size will be less then 5% of the total number of proteins, then this group would be dismissed. When the “Min. size of a subfamily” is set to 5% of the alignment size (i.e., the default) the algorithm tends to classify the dataset into several larger groups. If you set this parameter to "0" - meaning 3 proteins, i.e., the minimum number of proteins per subfamily possible - this setup would tend to classify the dataset into many smaller groups. We recommend to use the SSN viewer to iteratively adjust this parameter. E.g., the sequence similarity networks illustrated below were produced using the default setting for “Min. size of a subfamily”, i.e., 5%. You can see that proteins which seem to represent one large compact group were treated as outliers (i.e., colored in grey), as the size of that group was 4.5% from the total number of proteins:
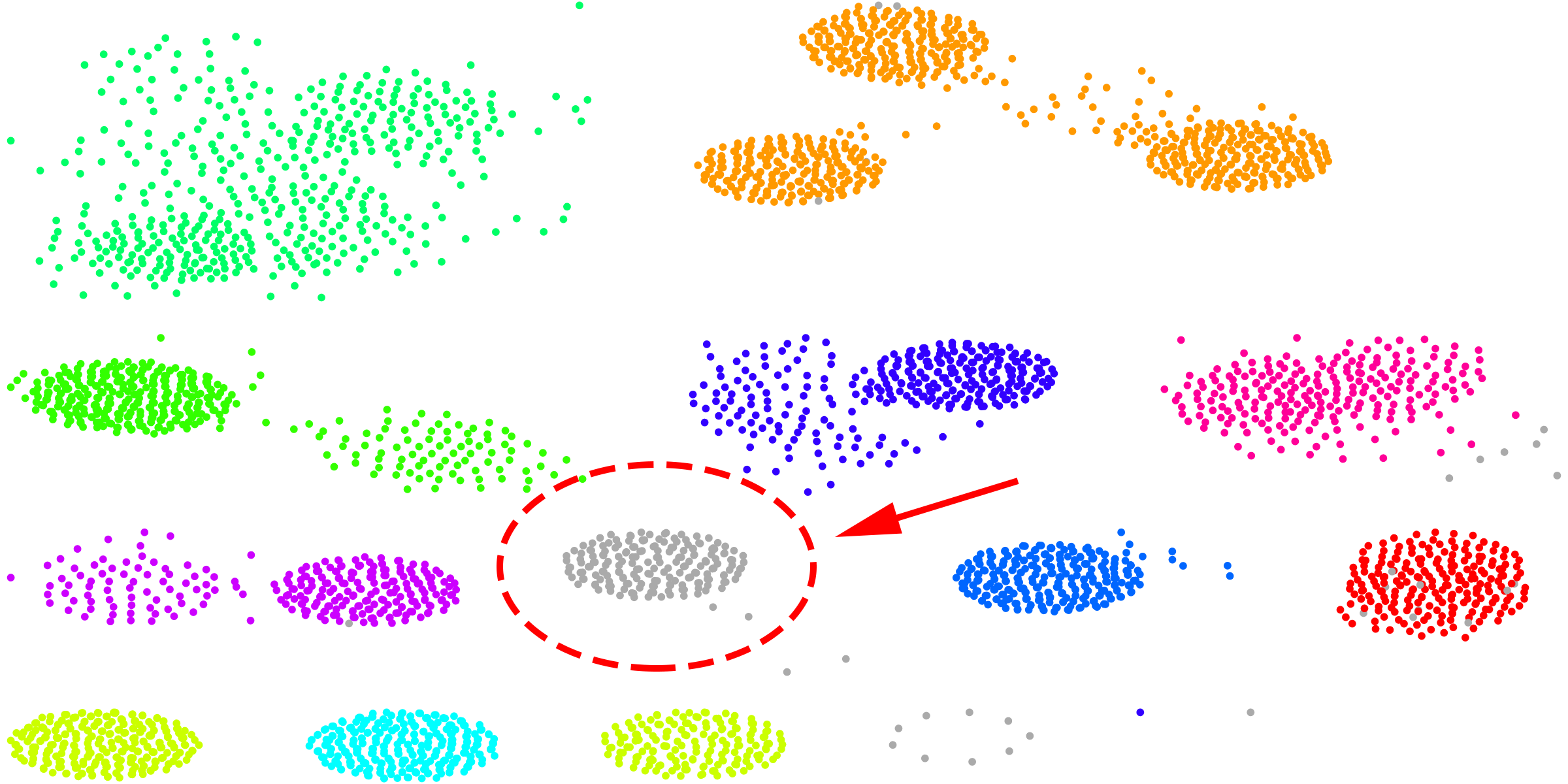
Running Zebra for the second time with the same input multiple alignment, but this time setting “Min. size of a subfamily” to 4% helped to preserve this data, i.e., as a result this group was treated as a separate subfamily to add value to sequence variability assessed by the bioinformatic analysis (see the Example 2 and the Demo mode for this example).
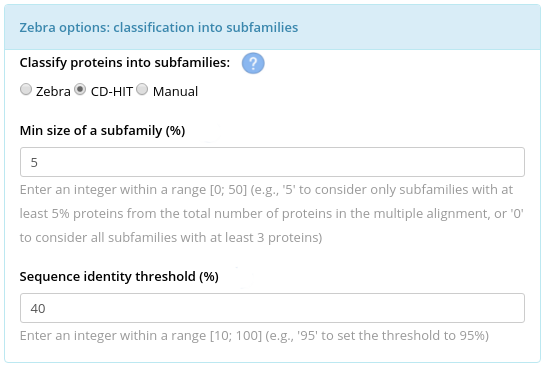
Alternatively, you can use CD-HIT to propose the classification automatically by "cutting the phylogenetic tree" corresponding to your input proteins at a selected level of sequence similarity. You would also need to set the “Min. size of a subfamily” parameter which in this case is qualitatively similar to what was previously discussed for the Zebra internal classification algorithm. Only one classification will be produced by the CD-HIT.
The third option would be to upload a file with explicit assignment of proteins to functional subfamilies. This custom classification can be created using a third-party algorithm, or guided by intuition and implementing manual curation. The format of the manual classification file is discussed in the section Input of the on-line documentation.