Preparing the input data for Zebra analysis
To use Zebra you have to submit (1) a multiple alignment of proteins in the FASTA format and (2) a representative protein (i.e., the query) structure in the PDB format which should correspond to one of the proteins in the multiple alignment. This input can be prepared automatically and fully on-line by the Mustguseal web-server. The Zebra2 currently supports multiple alignments of at most 16000 proteins. Sequence similarity networks would be generated if the input multiple alignment contains up to 8000 proteins.
- Overview of the input data
- Automatic preparation of the input data by Mustguseal
- Guidelines for manual preparation of the input data
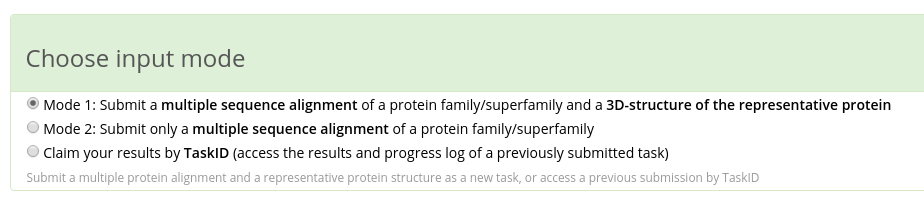
Zebra has two input modes:
- in Mode 1 you have to submit a multiple protein alignment in FASTA format and a representative protein (i.e., the query) structure in PDB format;
- in Mode 2 you can submit only a multiple alignment.
The details are provided below.
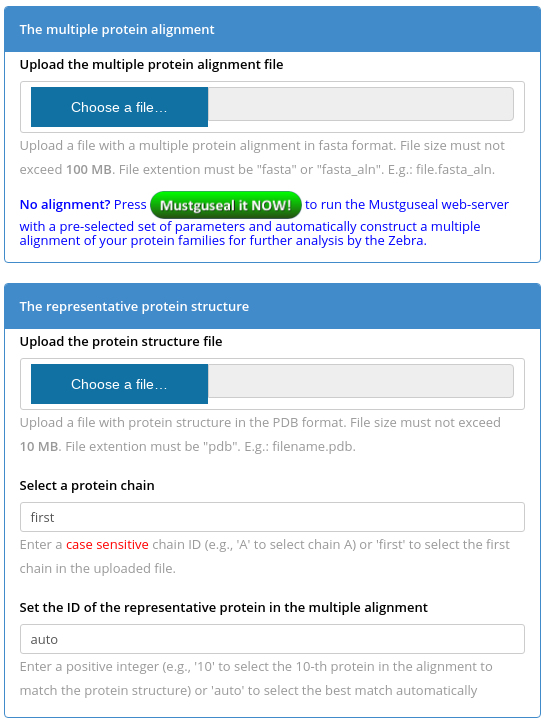
Mode 1. You have to submit two files to the Zebra server - a multiple protein alignment in FASTA format and a representative protein (i.e., the query) structure in PDB format. To select a protein chain for the bioinformatic analysis use the "Select a protein chain" field - you can type in a particular chainID (case sensitive) or leave it to "first" to select the first chain that appears in the PDB file. The representative protein structure should match one protein in the sequence alignment. You can set the "Set the ID of the representative protein in the multiple alignment" field to "auto" for the server to automatically match the PDB structure with the alignment to select the best pairwise sequence superimposition, or set a number corresponding to the order of appearance of the representative protein in the multiple sequence alignment file (the numbering starts from "1").
Mode 2. Alternatively, you can submit only a multiple protein alignment in FASTA format. In the Mode 2 only the sequence analysis of conserved and specific positions is performed, and all the 3D-based functionality is disabled (i.e., the 3D-mode, the 3D-viewer and on-line annotation of specific positions, etc.). In general, we recommend using the Mode 1 to facilitate further analysis of your protein family/superfamily.
Automatic preparation of the input data by Mustguseal
The input to the Zebra can be automatically prepared by the sister web-server Mustguseal via a web-interface. Mustguseal can automatically construct large structure-based sequence alignments of functionally diverse protein families that include thousands of proteins based on all available information about their structures and sequences in public databases. The alignment automatically constructed using that public web-server can then be automatically submitted to the Zebra web-server in one click.
Step 1: Choose the query protein. Choose the query protein based on your particular task and primary interest. It can be the target protein selected for the further experimental design, the most studied member of the superfamily, or a protein which you are the most familiar with.
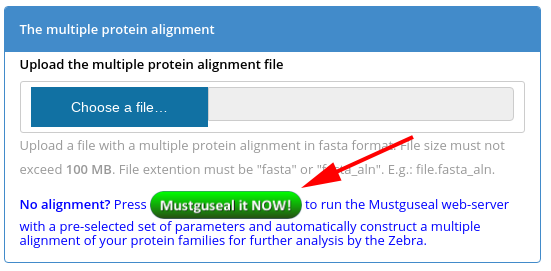
Step 2: Automatically construct a large alignment of diverse protein families. At the Zebra submission page press the "Mustguseal it NOW" button (see a screenshot below) or simply press this one -  . This will redirect you to the Mustguseal web-server and load a pre-selected set of parameters (i.e., the Scenario 2).
. This will redirect you to the Mustguseal web-server and load a pre-selected set of parameters (i.e., the Scenario 2).
Enter the PDB ID and chain ID of your query protein (e.g., PDB 3WGB chain A of L-allo-threonine aldolase from Aeromonas jandaei, as discussed here) in the "Query protein" box and press "Submit" to automatically construct a large structure-guided sequence alignment of proteins with high structural but low sequence similarity to your query protein.

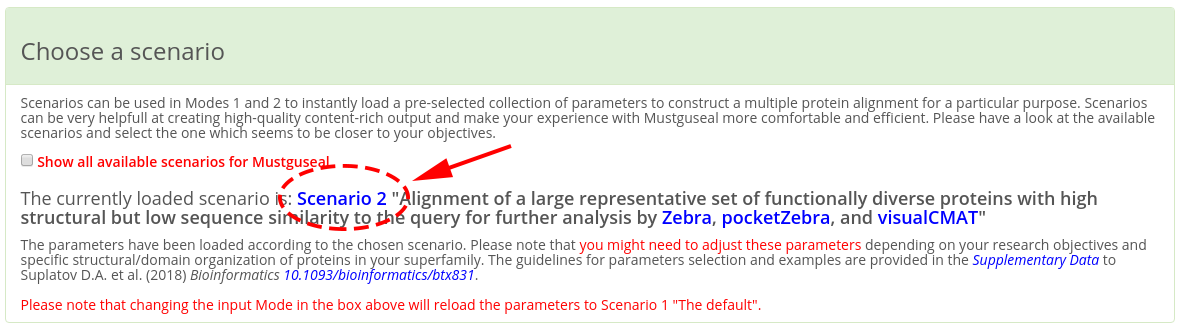
IMPORTANT! When making a submission to Mustguseal check that the "Choose a scenario" box states that the parameters have been set up according to the "Scenario 2" (see a screenshot below). If you have entered the Mustguseal submission page not from the Zebra web-page than other scenarios might be loaded. It that case set the "Scenario 2" manually by checking the corresponding radio button. The parameters in the "Scenario 2" were chosen to optimize the performance of Mustguseal at constructing large and diverse alignments of protein families for further analysis by Zebra. Please note that you might need to adjust these parameters depending on your research objectives and specific structural/domain organization of proteins in your superfamily. The guidelines for parameters selection and examples are provided in the Supplementary Data to the Mustguseal publication.
Step 3: Automatically transfer your data from Mustguseal to Zebra. A "Results" button will appear at the Mustguseal progress log page upon the completion of your task. Press that button to access the Mustguseal "Results" page. At that page page scroll down and press the "Submit to Zebra2" button. This will automatically transfer the collected sequence and structural data to the Zebra v.2 web-server.

Step 4: Start Zebra. Press "Submit" button to start the Zebra routine. When your task is processed, press "Results" to access the results and the download section, and press "Analysis" to load the interactive on-line analysis tools.
Using protein structures not (yet) in the PDB as queries to the Zebra and Mustguseal
Can a protein structure not yet in the PDB (or a homology model) be used as a query to the Zebra? Yes, it can. Upload the corresponding file in the PDB format to Zebra using the standard File Upload dialog box, and then upload the corresponding multiple alignment that contains this protein. If you do not have such an alignment - see below.
Can a protein structure not yet in the PDB (or a homology model) be used as a query to the Mustguseal to automatically construct the alignment for further processing by the Zebra? No, it can not. If your protein structure is new and not (yet) in the PDB (or a homology model) you have two options: (1) use a PDB of a close homolog to construct the alignment by Mustguseal and then upload the new protein structure in the PDB format together with the alignment to the Zebra (i.e., the PDBeFOLD web-server can be used to search for structurally similar proteins in the PDB to select such a homolog with a known 3D-structure) or (2) create and submit a custom-built core structural alignment to Mustguseal to build a corresponding structure-guided sequence alignment in Mode 2 (as explained here), and then submit it to the Zebra together with the new protein structure in the PDB format.
Guidelines for manual preparation of the input data
Alternatively, you can prepare the input manually. The multiple sequence alignment should represent the desired diversity among the protein families of interest (i.e., contain homologous proteins with different function, stability and regulation). The representative protein structure is expected to correspond to a protein in the sequence alignment. Choose the representative protein based on your particular task and primary interest. It can be the target protein selected for the further experimental design, the most studied member of the superfamily, or a protein which you are the most familiar with. You should always aim at submitting a representative PDB structure that corresponds to a protein in the alignment with at least 95% pairwise sequence identity. If structural information is not available for all proteins in your alignment then you could use the structure of a very close homolog from the PDB database or build a 3D model of the representative protein based on the available structural data using the homology modeling (e.g., with the help of the highly capable Modeller software).
Protein naming scheme For multiple alignments created by the user (i.e., without the Mustguseal), the correct format of protein names for compatibility with the Zebra on-line analysis tools is responsibility of the user. The format of protein names (i.e., sequence titles in the FASTA alignment file) that can be recognized by the Zebra is explained below. Please comply with this input format to activate the detailed annotation of proteins with links to the external resources (i.e., PDB, Uniprot, BacDive, BRENDA) at the Zebra analysis page (see Results for details)
The PDB codes should be provided to the Zebra in the following format within the protein name in the multiple alignment FASTA file: [ID]_[PDBC]_[chain], where [ID] is any number, [PDBC] is the 4-digit PDB code, and [chain] is a 1-character chain identifier, e.g.:
>122_3tec_E
YTP-N-DPY-FSSRQYGPQKIQAP
QAWDIAEGSG--AKIAIVDTGVQS
NHP---DLAGKVV-----------
The UniProt AC codes should be provided to the Zebra in the following format within the protein name in the multiple alignment FASTA file: [AC]=[anyinfo], where [AC] is the UniProt AC code containing 6-to-10 characters, e.g.: >P08594=AQL1_THEAQ_Aqualysin1_OS=Thermus_aquaticus_OX=271_GN=pstI_PE=1_SV=2
QSPA---PWGLDRIDQRDLPLSNS-Y
TYTATGRGVNVYVID--TGIRTTHRE
FGGRARV-----GYDALGG-------
General requirements for the input files
The input multiple alignment:
- should contain protein amino acid sequences;
- should contain at least six proteins;
- should be an alignment (i.e., not just sequences, but aligned sequences, i.e., "sequences with gaps");
- the "-" character should be used for a gap;
- the special characters in the protein names are not allowed and will be automatically substituted for "_";
- the very long protein names will automatically truncated to the first 100 characters;
- the special characters in the protein sequences are not allowed and will be automatically substituted for gaps;
- should be in the FASTA format (not ClustalW, not Phylip, etc.). If you do not know what is the format of your alignment - submit it to Zebra and you`ll find out. If you have your alignment in the wrong format use a sequence format converter, e.g., sequenceconversion.bugaco.com;
- The policy on the non-standard amino acid codes is as follows: the non-standard amino acid codes are automatically converted to a predefined standard code, i.e., B → D, J → I, X → G, Z → E, then U (i.e., selenocysteine) is converted to C, and O (i.e., pyrrolysine) is converted to K.
The input protein structure:
- should contain the coordinates of amino acids atoms of one protein;
- should be in the PDB format;
- should correspond to (i.e., ideally should be 100% identical to) one protein sequence in the multiple alignment;
- may not be 100% identical to any protein sequence in the multiple alignment. The preprocessing script will automatically select the representative sequence from the multiple alignment by the best pairwise match between your PDB structure and any sequence in the alignment. All inconsistencies between the representative protein structure and sequence will be removed. You should always aim at submitting a representative PDB structure that corresponds to a protein in the alignment with at least 95% pairwise sequence identity. You will be allowed to proceed with up to 50% sequence similarity between the two, however, this may cause errors during the bioinformatic analysis.
- should contain all chains of the biological unit (e.g., A, B, C) even if the multiple alignment contains the sequences of only one chain (e.g., A);
- may contain heteroatoms. All non-protein atoms (e.g., of a substrate) should have the HETATM prefix in the PDB file. Non-canonical amino acids will be automatically changed to the canonical equivalents (i.e., SME/MSE to MET). Ligands, cofactors, solvent and other instances should be in the HETATM field and will not be used for the bioinformatic analysis but will be used to prepare the graphical output and can help with the interpretation of functional and regulatory significance of the predicted co-evolving positions.
- each item in the PDB (amino acid or a ligand) should have a unique identification (chain id + residue name + residue ID);
The manual classification file
To identify the specific positions in the multiple alignment the input set of proteins has to be classified into functional families/subfamilies. The classification can be performed automatically (see Parameters for details) or provided by the user. The manual classification file has to be uploaded at the "Zebra options: classification into subfamilies" panel (choose "Manual", see the figure above). The manual classification file:
- is a plain text file;
- it contains only numbers;
- it includes all numbers from 1 to N, corresponding to the protein rank in the alignment file, where N is the number of proteins in the alignment (i.e., the rank of the last protein, starting from 1). I.e., outliers are not allowed in the manual classification file;
- the file should contain at least two lines of numbers corresponding to proteins in the alignment, where each line defines a group/subfamily;
Example of a manual classification file:
1 2 3 4
5 6 7 8 9 10 11 12 13 14 15 16 17 18
This file classifies a multiple alignment of 18 proteins into two subfamilies, of 4 and 14 proteins, respectively, i.e., the first four proteins comprise the first subfamily, the next 14 proteins - the second subfamily.
You should know that Zebra actually prints all intermediate multiple alignment files and corresponding classification files that are created during the automatic classification process. These files are not listed in the Download section of the results, but instead appear in the Log file at the end of "Step 4: The Zebra bioinformatic analysis", e.g.:

These files can be downloaded and used to run Zebra with a different set of parameters and only one selected classification, but also as templates to prepare manual classification.