Zebra3D Input
The input to the program is (1) a folder with aligned protein 3D-structures presented as separate files in the PDB format; (2) a file with the corresponding sequence representation of 3D-alignment in the FASTA format. The program itself does not impose limitations on the number of input PDB structures or their dimensions. However, analysis of a larger number of aligned PDB structures would require more time and resources to complete. The required input to Zebra3D can be automatically prepared and subjected to the Zebra3D analysis with the default settings fully on-line using the Mustguseal web-server in Mode 4. To simplify the preparation of the input data, separate chains of heteromeric proteins should be evaluated as independent tasks.
- Overview of the input data
- Automatic preparation of the input data by Mustguseal and on-line analysis by Zebra3D with default settings
- Guidelines for manual preparation of the input data
Overview of the input data
The input to the program is a 3D-alignment of protein structures presented as (1) a folder with separate PDB entries corresponding to the aligned proteins in the PDB format, accompanied by (2) a file with sequence representation of the 3D-alignment in the FASTA format. The respective input data should look like this in your file manager:
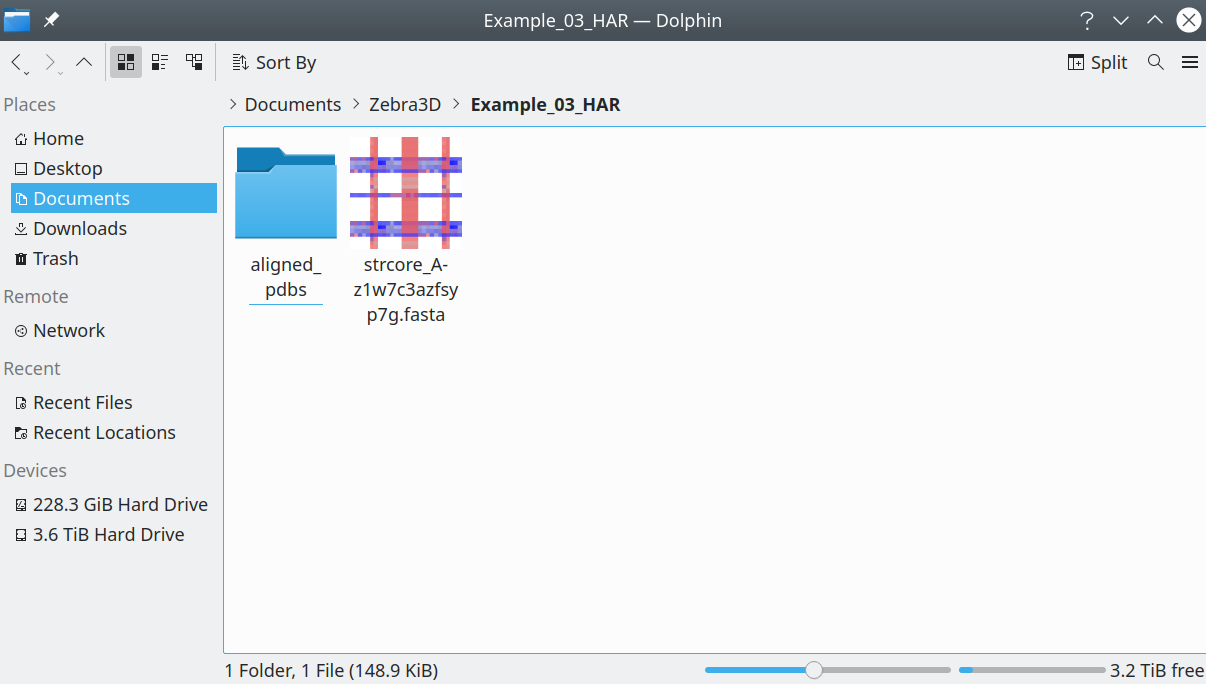
[Click here to enlarge]
The protein 3D-structures should be presented as separate files in the PDB format:
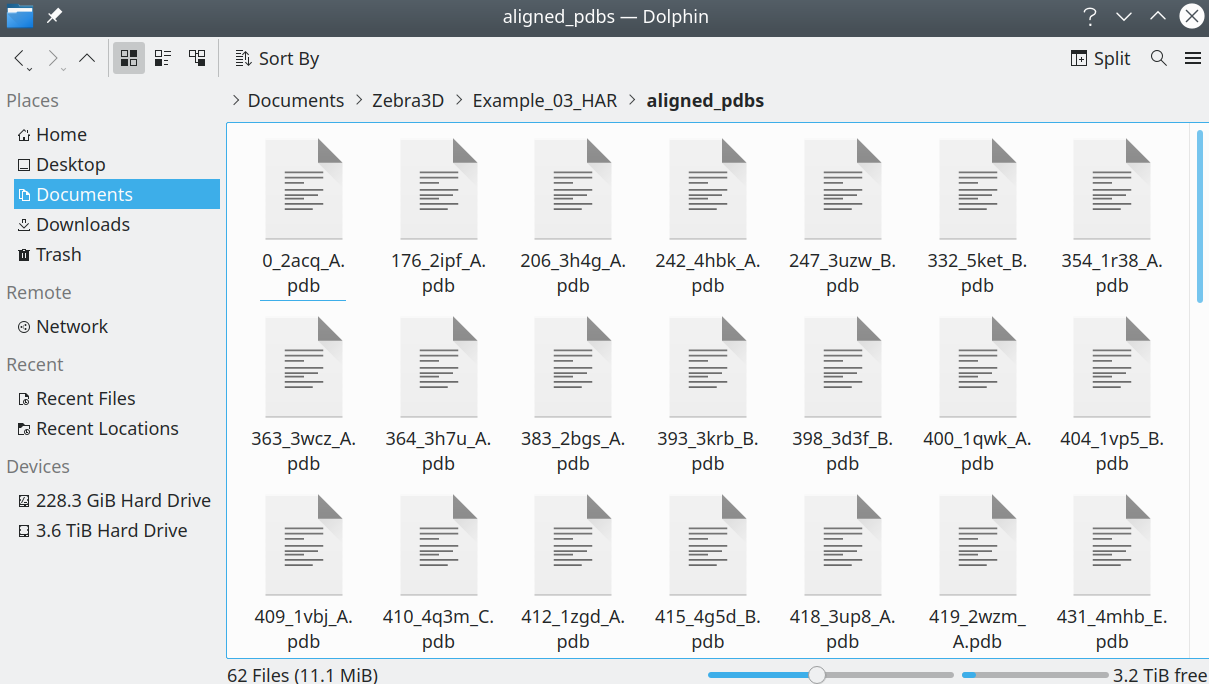
[Click here to enlarge]
These separate PDB files should preserve the common coordinate space, i.e. they should be actually aligned with each other. If opened all at once in PyMol, the viewport should reveal a biologically meaningful 3D-superimposition:
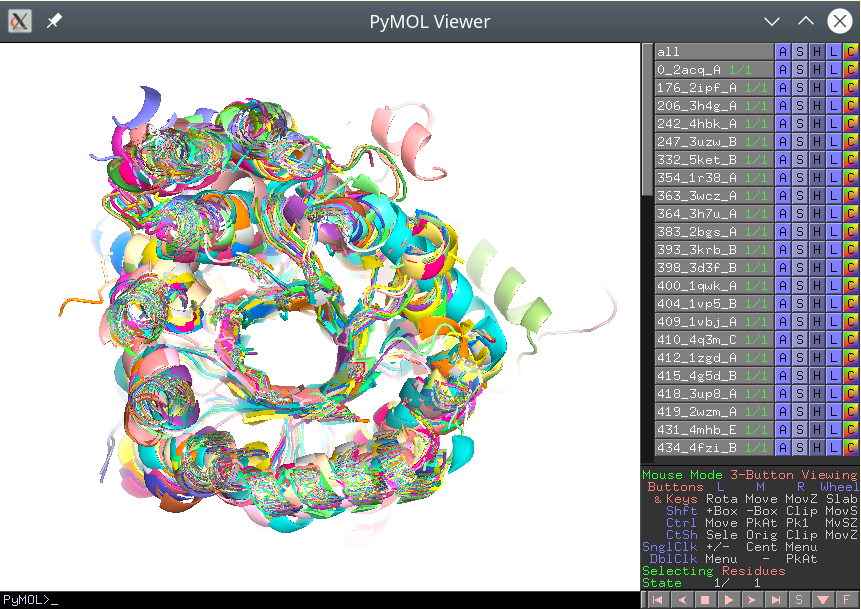
[Click here to enlarge]
The accompanying file with sequence representation of the 3D-alignment should be a plain text file in the FASTA protein alignment format:
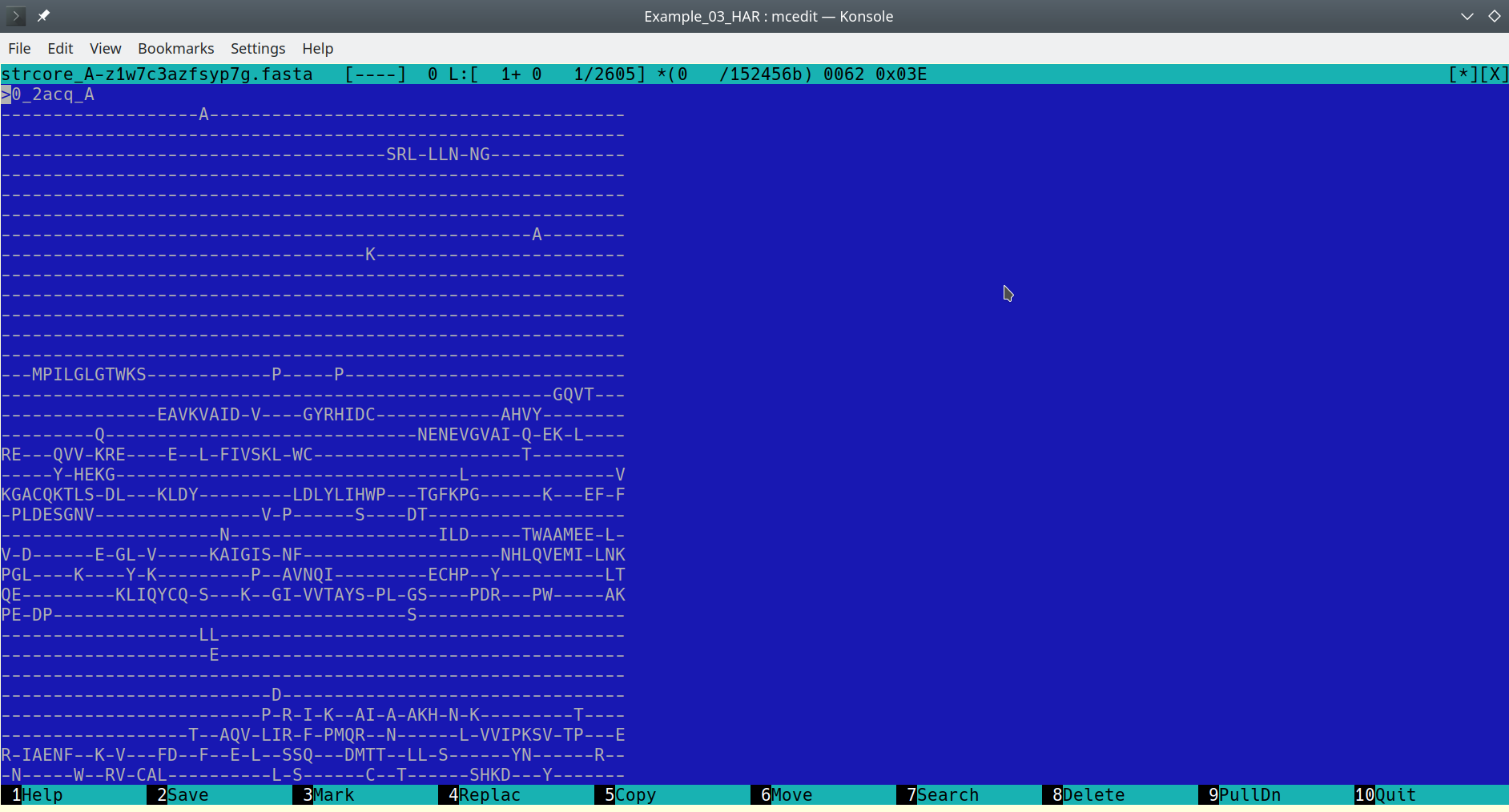
FASTA alignment file opened in a text editor [Click here to enlarge]
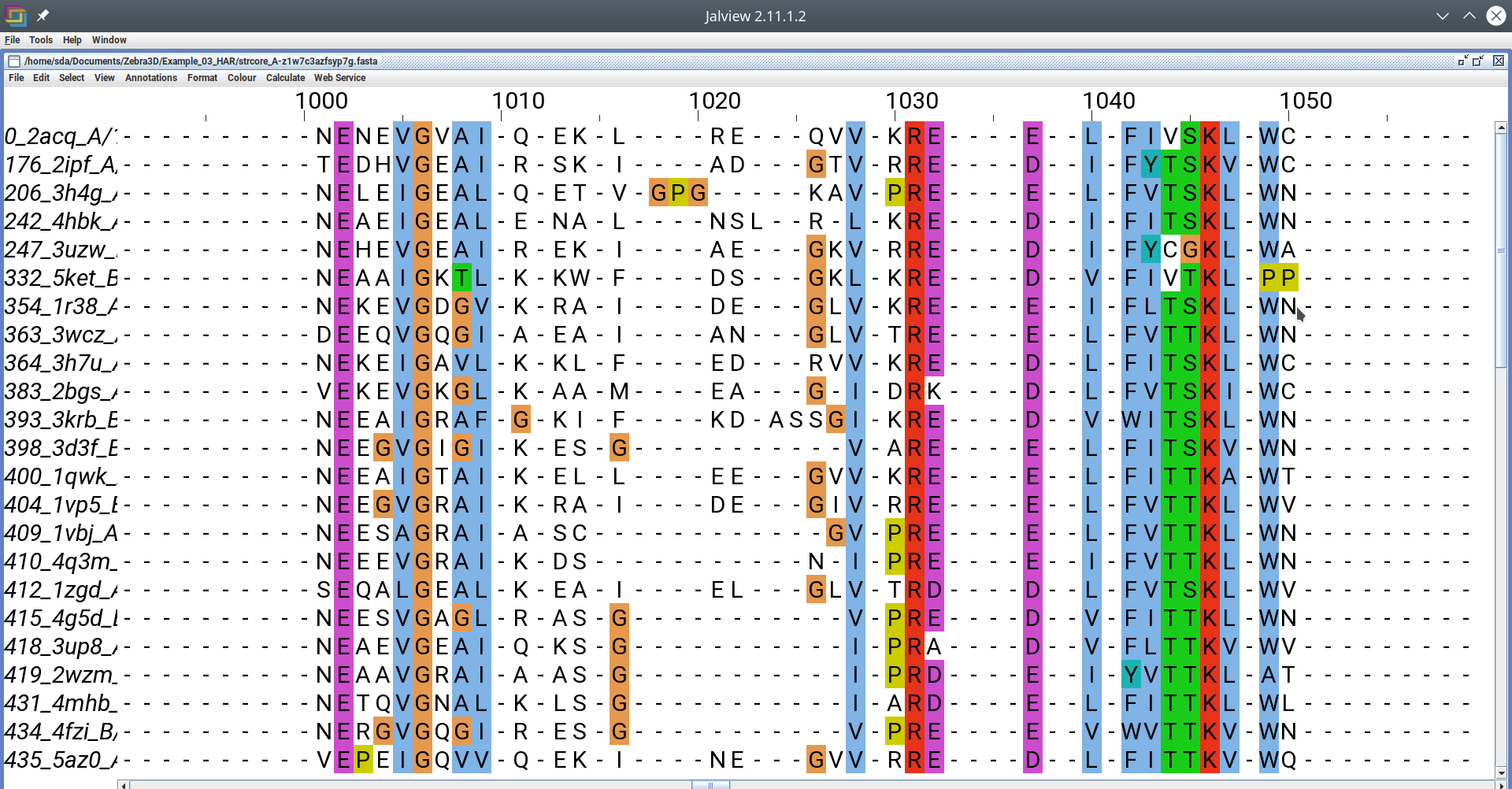
FASTA alignment file opened in Jalview editor [Click here to enlarge]
Automatic preparation of the input data by Mustguseal and on-line analysis by Zebra3D with default settings
The Mustguseal web-server in Mode 4 can be used to (1) collect and superimpose a diverse set of protein 3D-structure (i.e., prepare the input 3D-alignment automatically) and then to (2) run the Zebra3D analysis with the default setup on-line. The user can then download both the automatically constructed 3D-alignment and the default Zebra3D results from that web-server. The Zebra3D results can be analyzed straightaway. The 3D-alignment can be used as input to a local installation of Zebra3D to further improve the bioinformatic analysis by fine-tuning the parameters. The details are provided below.
The collection and subsequent 3D-alignment of diverse homologs can be handled fully on-line by the Mustguseal web-server. Starting from the selected query protein (submitted to the web-server as a PDB code and chain ID), the Mustguseal will collect and align a non-redundant set of protein 3D-structures from the PDB database sharing a certain degree of similarity with the query. The 3D-similarity is defined by the percentage of secondary structure equivalences between the query protein and each target in the PDB, according to the user-defined thresholds. To take advantage of that web-method, submit a PDB code of the query protein in the “Mode 4”. You can learn more about the Mustguseal by reading the dedicated on-line tutorial, starting from this page.
You can simply press this button for an automatic redirect  , or you can go to the Mustguseal main page and manually select the "Mode 4" input mode. Enter the PDB code and chain ID of the selected query protein, and press "Submit" to start the automatic alignment construction process:
, or you can go to the Mustguseal main page and manually select the "Mode 4" input mode. Enter the PDB code and chain ID of the selected query protein, and press "Submit" to start the automatic alignment construction process:
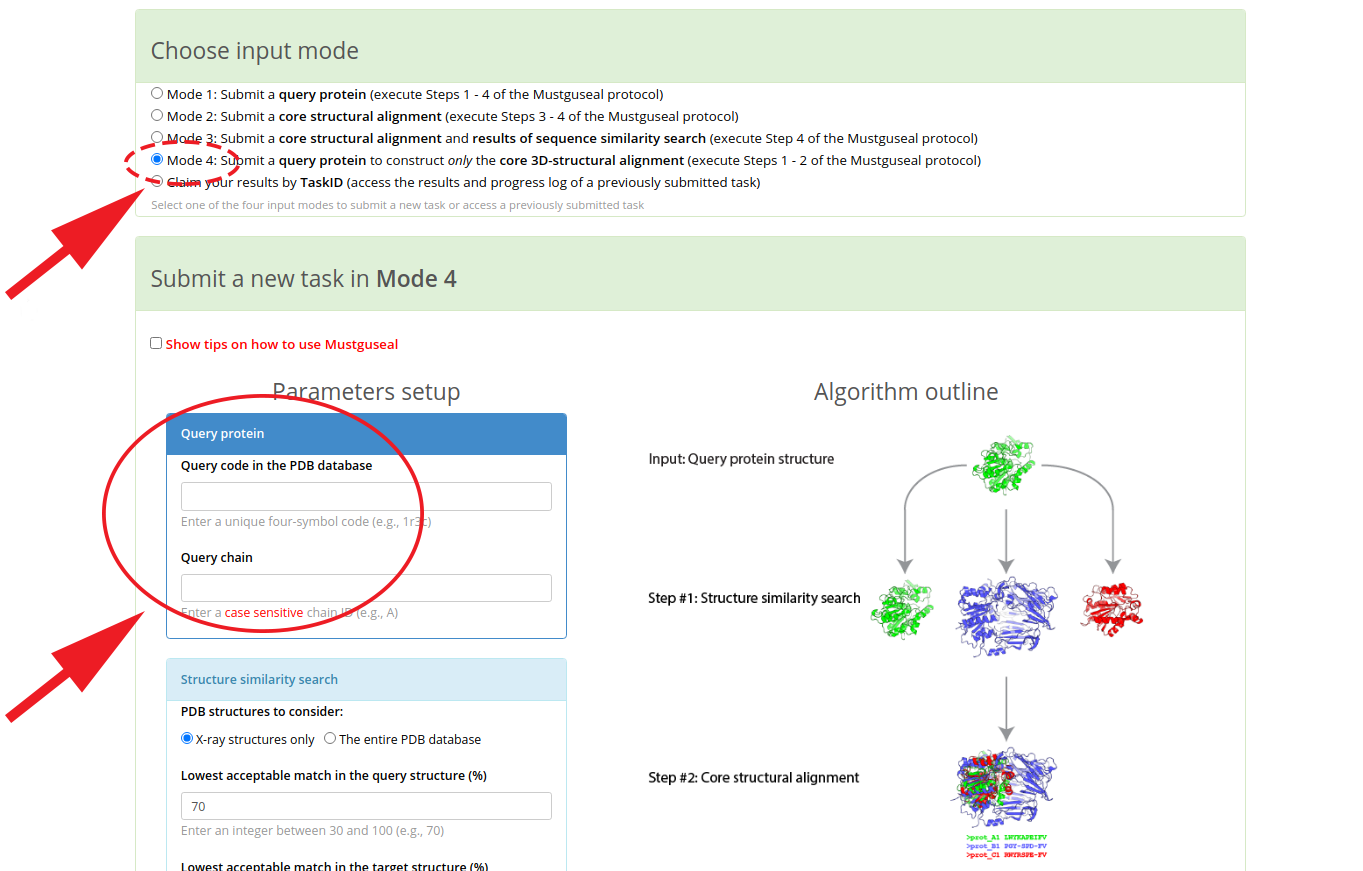
[Click here to enlarge]
The default results of Zebra3D will be provided at the “Results” page in the "Supplementary output" section as “Download the Zebra3D results”; the guide to Zebra3D results is provided on a dedicated page:
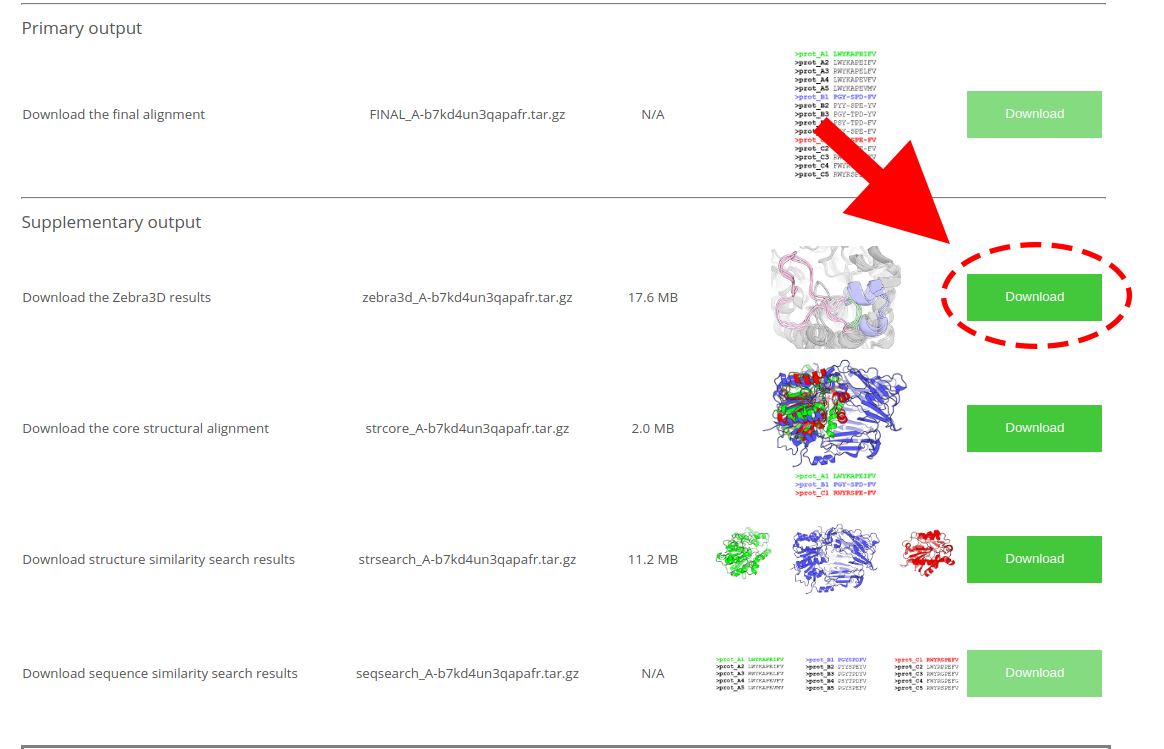
[Click here to enlarge]
The collected and superimposed 3D-entries and corresponding sequence representation of the resulting 3D-alignment in the FASTA format can be acquired at the “Results” page using the “Download the core structural alignment” button:

[Click here to enlarge]
The respective archive package should be downloaded to the user’s computer and unpacked to reveal the data to be used as input to Zebra3D: (1) the folder with superimposed PDB entries entitled aligned_pdbs and (2) the corresponding FASTA file with the sequence representation entitled strcore_TaskID.fasta:

[Click here to enlarge]
You might also be interested in downloading the file with a detailed annotation of each PDB entry presented in a conveniently organized table (i.e. featuring annotation embedded into each PDB file). You can download the corresponding archive at the Mustguseal "Results" page using the “Download structure similarity search results” button:
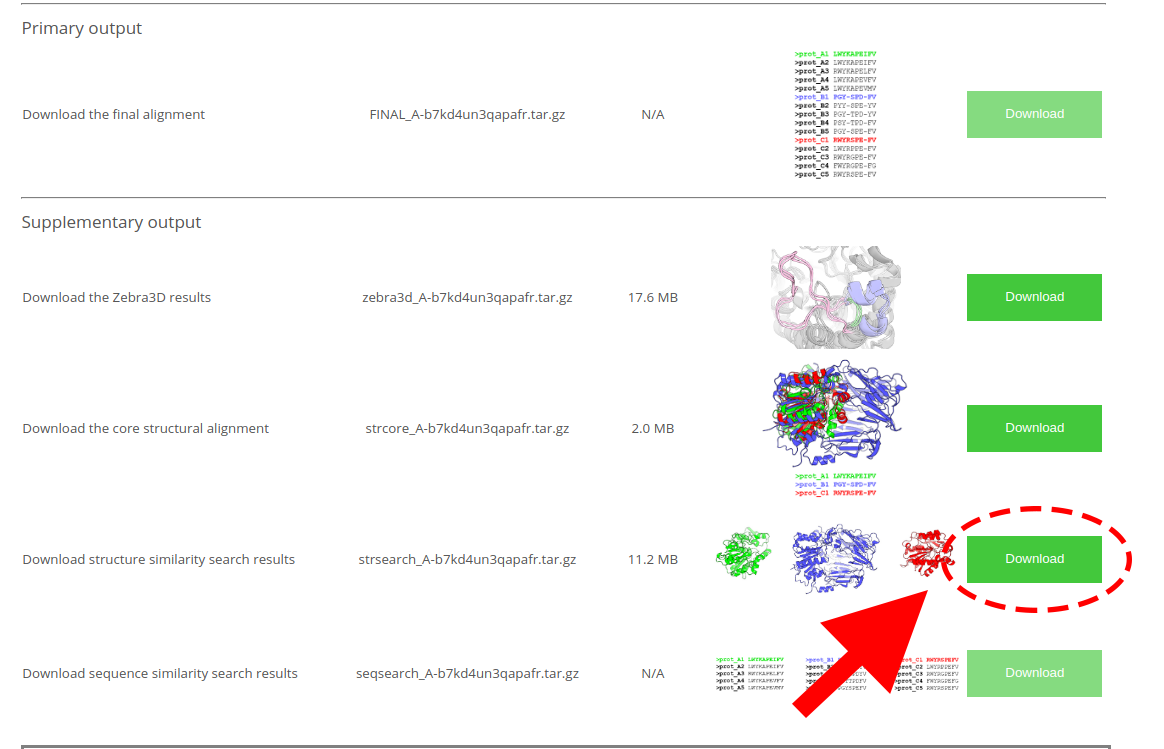
[Click here to enlarge]
The archive with data should be downloaded to the user’s computer and unpacked to reveal the superimpose.list file:
[Click here to enlarge]
The superimpose.list is a plain text file, which may be opened using any text viewer/editor of your choice, and contains the complete list of all PDB entries that were found to be structurally similar to your query. Those PDB entries that were finally selected for the 3D-alignment will be marked by the '*' sign, as explained below in more details. For each PDB entry, which was collected by the Mustguseal as being structurally similar to your query, the file contains a detailed annotation which is conveniently organized in a form of a table:
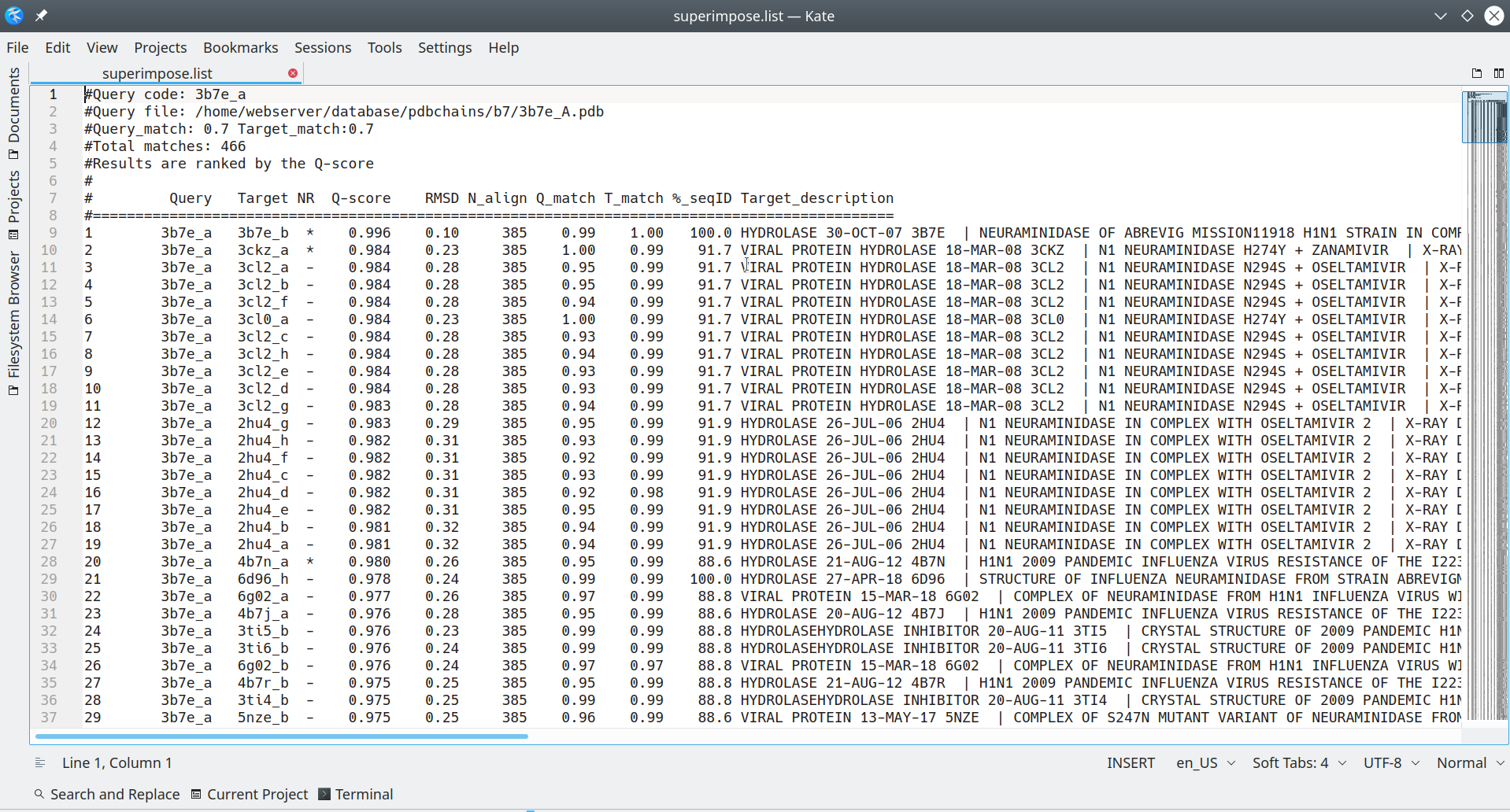
[Click here to enlarge]
By default, the Mustguseal runs the 3D-structure similarity search versus the PDB database to collect all PDB entries which are structurally similar to the query protein (given the selected 3D-similarity thresholds). Then, the Mustguseal attempts to automatically select a non-redundant subset of no more than 64 entries at the 95-40% pairwise sequence identity threshold for the "core collection". I.e. if the 95%-non-redundant subset contains more than 64 proteins, then the 90% threshold will be probed, then 85%, etc. The finally selected non-redundant core collection is further subjected to the 3D-alignment. In the superimpose.list file, the proteins which were automatically selected at the 95% pairwise sequence identity threshold are marked by the '*' sign in the 'NR' (i.e., non-redundant) column. I.e., all PDB entries being structurally similar to your query will be listed in that file, and the 95%-nr set will be marked by the '*' sign. The 95% threshold may not be the one actually used to construct your particular 3D-alignment. You may need to view the on-line log file for the Mustguseal task to learn the finally selected threshold, e.g.:
Info: Selecting a non-redundant set of structures for the core structural alignment
Info: A non-redundant set of structures clustered at the 95% level of sequence identity has 150 proteins
Info: A non-redundant set of structures clustered at the 90% level of sequence identity has 102 proteins
Info: A non-redundant set of structures clustered at the 85% level of sequence identity has 81 proteins
Info: A non-redundant set of structures clustered at the 80% level of sequence identity has 62 proteins
Info: A set of 63 proteins (the non-redundant set + the query) has been selected for the core structural alignment
You should then look for the corresponding file superimpose.sXX.list in the downloaded archive, e.g. superimpose.s80.list will contain the 80%-non-redundant subset marked by the '*' sign in the 'NR' (i.e., non-redundant) column.
Guidelines for manual preparation of the input data
You should take the following notes when preparing the input, as explained below. These rules are fulfilled by the Mustguseal web-server when constructing the alignment automatically. If you are having difficulties with preparing your input data set for Zebra3D, you should first run Mustguseal with some query and settings, and study the format of the automatically prepared data. Would you still have problems with launching Zebra3D, don't hesitate to send us a question.
- As mentioned many times before, the PDB files which are submitted to the Zebra3D should preserve the common coordinate space, i.e. they should all be aligned to each other. If, by mistake, you submit the raw (unaligned) PDB files to the Zebra3D, the program will warn you of significant deviations between the protein structures (exceeding 9 angstroms on average per any two PDB entries), but may still go on with the analysis which would most likely produce meaningless output;
- Protein names in the sequence alignment should exactly correspond to the names of the respective PDB files. Minor inconsistencies could be corrected by the program automatically, and the corresponding warning messages will appear in the program output. In case of significant differences, the program will terminate with an error message;
- For example, if these are the names in the FASTA file ... :
>0_1bvt_A
>17_6dja_A
>60_1a7t_A
>72_5n58_B
>73_6lf4_D
>81_2y8b_A
>88_5lca_A
... then the names of the respective PDB files should be like this:
0_1bvt_A.pdb
17_6dja_A.pdb
60_1a7t_A.pdb
72_5n58_B.pdb
73_6lf4_D.pdb
81_2y8b_A.pdb
88_5lca_A.pdb - The folder with PDB files should contain only the entries mentioned in the FASTA sequence alignment, and vise versa;
- The PDB files and FASTA file should only contain data which is meant for the Zebra3D analysis. E.g. water molecular should be removed from the PDB files and should not appear in the sequence version as "X" residues. Mustguseal web-server does such "cleaning" automatically. Generally speaking, preserving heteroatoms in PDB structures should not be a problem in any case, as Zebra3D considers only the backbone atoms; however, handling of unusual/unnecessary content in the PDB/FASTA files was not tested;
- The PDB entries should be as complete as possible. You may attempt to reconstruct/model missing loop segments prior to submitting the task to Zebra3D. The program will process incomplete segments as they are, i.e. only the actually available coordinates will be included in the analysis, scoring and ranking.
The user-curated collection of proteins of interest can be aligned using any bioinformatic software for 3D-alignment that produces both the PDB- and sequence-versions of the output superimposition. In particular, we have recently introduced parMATT – the first parallel re-implementation of the highly successful MATT algorithm to align multiple protein 3D-structures by allowing translations and twists (10.1093/bioinformatics/btz224). The parMATT is faster - i.e. it provides an opportunity to accelerate a 3D-alignment even on a single multi-core CPU, but its key advantage is the ability to run on distributed-memory systems, i.e. computing clusters and supercomputers hosting memory-independent computing nodes. The source code and user manual are available on-line at https://biokinet.belozersky.msu.ru/parMATT. When using parMATT to prepare the input to Zebra3D, you should activate the “Partial alignments” postprocessing step by specifying the “-p1” command line flag (as explained in the corresponding on-line manual).