Guide to pocketZebra output
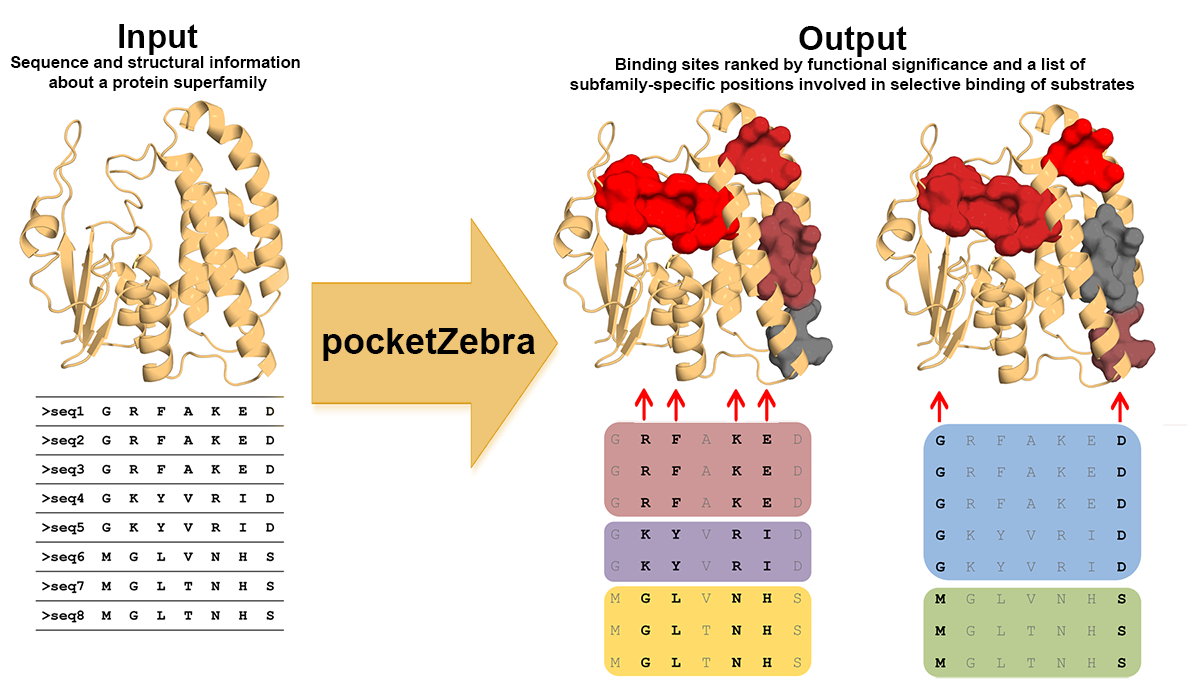
Output description
This page describes the output produced by pocketZebra server. See the Examples page for some real output data.
The pocketZebra output page has the following sections:
- The Header
- Functional Subfamily Classifications
- Functional Sites
- Interactive 3D structure viewer
- Subfamily specific positions
- Download section
The Header

 |
Job Id access code is a unique identifier of your submission. JobID can be shared with a colleague and used to access the results at any time. |
 |
View Log button. Access the full log of pocketZebra to understand the workflow, review errors and warning messages. |
 |
Delete Job button. Irreversibly deletes your results from the server. |
Functional Subfamily Classifications

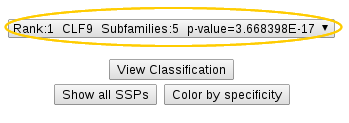 |
Drop-down list shows the proposed classifications of proteins from your input alignment into functional subfamilies. Each classification is described by its rank, identifier, number of subfamilies and a corresponding P-value. Classifications are ranked by declined significance and the first most significant classification is shown by default.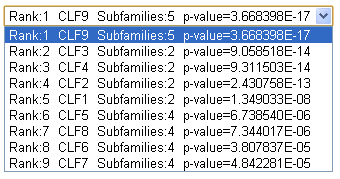 |
 |
View Classification button will bring up a page with a list of proteins from your input alignment organized into functional subfamilies. An example of a two-subfamilies classification is shown below: the first and second subfamilies (SUBF1 and SUBF2) of the classification with identifier CLF6 are shown.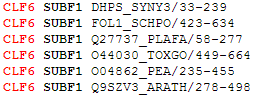  |
 |
Show all SSP's button allows you to visualize (see Visualisation section below) all significant subfamily specific positions identified for the selected classification.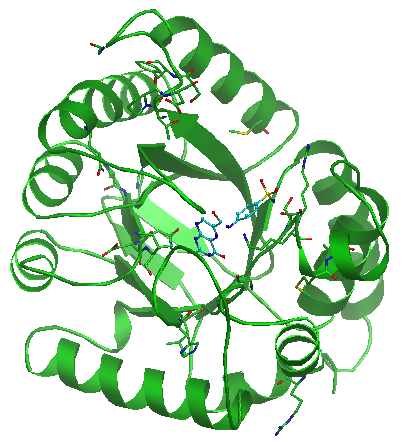 |
 |
Color by specificity button allows you to color CA atoms of all subfamily specific positions (see Visualisation section below) identified for the selected classification. The positions are gradient-painted according to calculated specificity Z-scores: red stands for highly significant hits, green – for non-specific positions.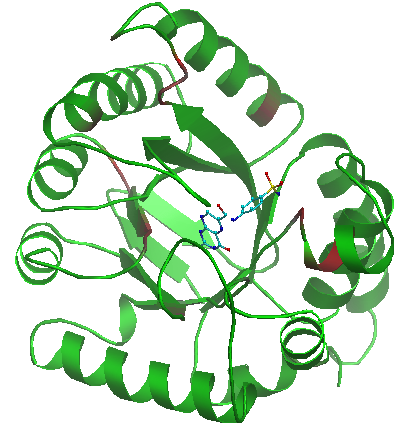 |
Functional Sites

 |
Drop-down list contains subfamily-specific binding sites (pockets) ranked in declined significance. Each item is described by the rank, name of the pocket and a corresponding P-value. The first most significant pocket is shown by default as mesh (see Visualisation section below). |
 |
Surface checkbox. Use this checkbox to visualize the selected pocket as solid surface.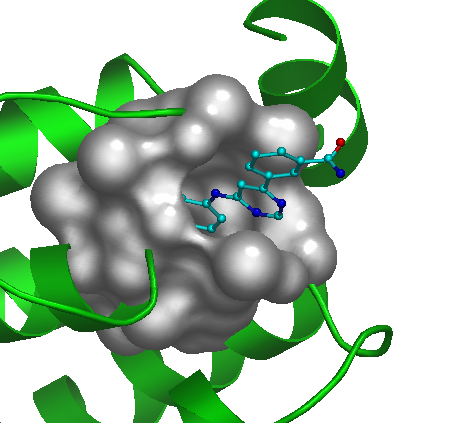 |
 |
Envelope checkbox. Use this checkbox to visualize the selected pocket as mesh.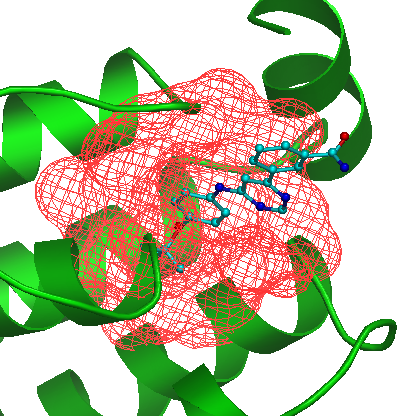 |
 |
Spheres checkbox. Use this checkbox to visualize all atoms of the pocket as "spheres".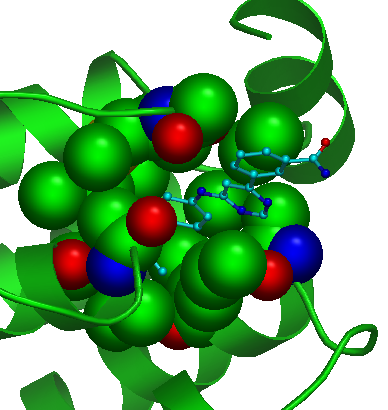 |
 |
Sticks checkbox. Use this checkbox to visualize all amino acids of the pocket as "sticks".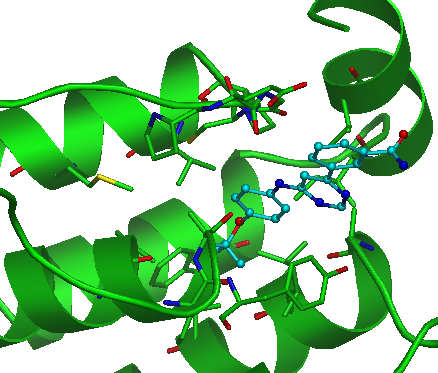 |
Interactive 3D structure viewer
pocketZebra implements JavaScript and Java-based openAstexViewer plugin for on-site visualization of the results. In order to enable this feature you will have to approve execution of the Java plugin - press "Run" (or "Proceed") button when prompted to start the application (Windows Example1, Windows Example2, Linux Example). Read more about managing typical Java security issues
{kind=link}
{kind=link}
{kind=link}
While pocketZebra output is primarily web-based and viewable on the website, the off-line version of the output is also available. Therefore, if you do not want to use the plugin you can still use pocketZebra and download the results to your computer for off-line use.
The view of the loaded molecule can be controlled using the left mouse button, while the right mouse button is reserved for bringing up the popup menu. Detailed description of OpenAstexViewer control and interface can be found at the original site.
By default the amino acid chain of an uploaded molecule is rendered in the "cartoon" representation (green color), hetero atoms are shown as "sticks" (cyan) and waters, ions etc. are hidden. The most relevant pocket is automatically visualized using envelope option (see Functional Sites section):
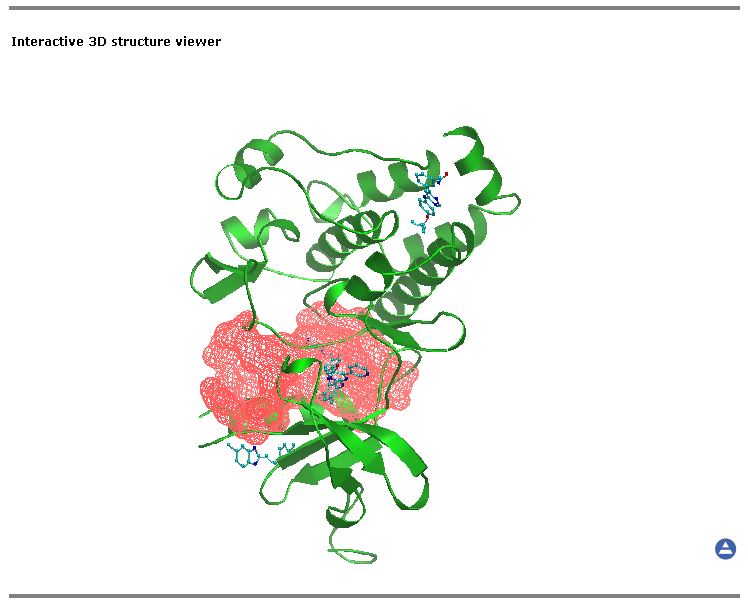
The settings you make in other sections of the output page (see Functional Subfamily Classifications, Functional Sites, Subfamily specific positions sections) will affect the 3D representation.
Subfamily specific positions
This section describes subfamily-specific positions located within a selected pocket: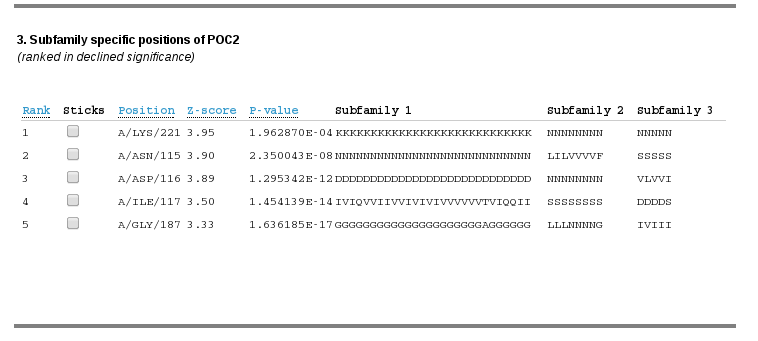
The data is presented as a table, each row corresponds to a subfamily-specific positions and each columns corresponds to the following data for each position:
| Rank | Statistical significance rank of a subfamily-specific position, smaller ranks indicate higher specificity. Subfamily-specific positions are ranked in declined significance. |
| Sticks | Use the checkbox to visualize the amino acid on the representative PDB structure as "sticks". |
| Position | Chain, residue name and residue identifier that corresponds to the subfamily-specific position in the representative PDB structure. |
| Z-score | Specificity Z-score. Larger Z-scores indicate higher specificity of a column of a multiple alignment. |
| P-value | Statistical significance of an observed set of first k subfamily-specific positions, where k is the rank of the current position from the list. P-value for a position with rank k is a probability for a set of first k scores to be observed by chance. Select the lowest global P-value to get a set of the most significant subfamily-specific positions for further evaluation. |
| Subfamily 1, Subfamily 2 etc. | Amino acid content of the corresponding column of a multiple sequence alignment classified into functional subfamilies. |
Download section
This section contains the off-line version of the output:
 |
The structure provided to the server is used to automatically produce PyMol session files (.pse) with structural representations of subfamily-specific positions within identified pockets for each subfamily classification. These files can be downloaded and used straightaway for structural analysis with PyMol without any additional programming experience. .pse files are organized as follows:
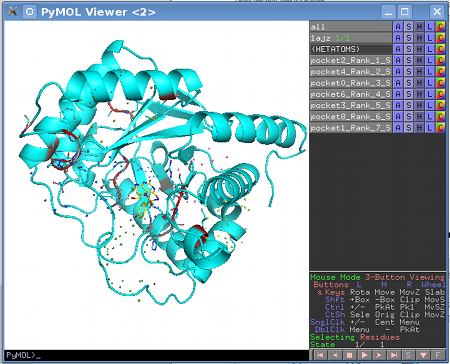 |
 |
pocketZebra text output file. It is a plain text file which contains context-specific headers at the beginning of each line for easy parsing using command-line tools and simple editors and can thus be integrated into automatic pipe-lines. Detailed information on pocketZebra output can be found below** |
 |
Download Bioinformatic analysis output file. Detailed information on Bioinformatic analysis output can be found here. |
**pocketZebra output file details
The output file is partitioned by blocks, each containing information for a particular functional subfamily classification.The head of a block contains a classification ID (CLF6), statistical significance rank and a P-value (outlined):

The block starts with the description of identified subfamilies as described in Functional Subfamily Classifications section:

followed by the ranked pocket blocks with subfamily-specific positions:
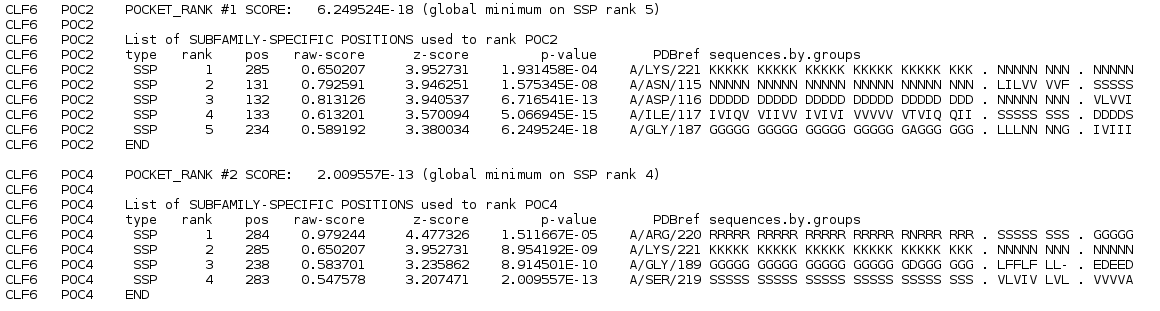
The table of subfamily-specific positions is similar to one in the Subfamily specific positions section:
| Rank | Statistical significance rank of a subfamily-specific position, smaller ranks indicate higher specificity. Subfamily-specific positions are ranked in declined significance. |
| Pos | Index of a column in the multiple alignment. |
| Raw-score | Specificity score according to original Specificity scoring function as described in Algorithm section. |
| Z-score | Specificity Z-score. Larger Z-scores indicate higher specificity of a column of a multiple alignment. |
| P-value | Statistical significance of an observed set of first k subfamily-specific positions, where k is the rank of the current position from the list. P-value for a position with rank k is a probability for a set of first k scores to be observed by chance. Select the lowest global P-value to get a set of the most significant subfamily-specific positions for further evaluation. |
| Reference | Chain, residue name and residue identifier that corresponds to the subfamily-specific position in the representative PDB structure. |
| PDBref | Corresponding residue name in a reference sequence. |
| Sequence.by.group | Amino acid content of the corresponding column of a multiple sequence alignment classified into functional subfamilies. |