Guide to pocketZebra input
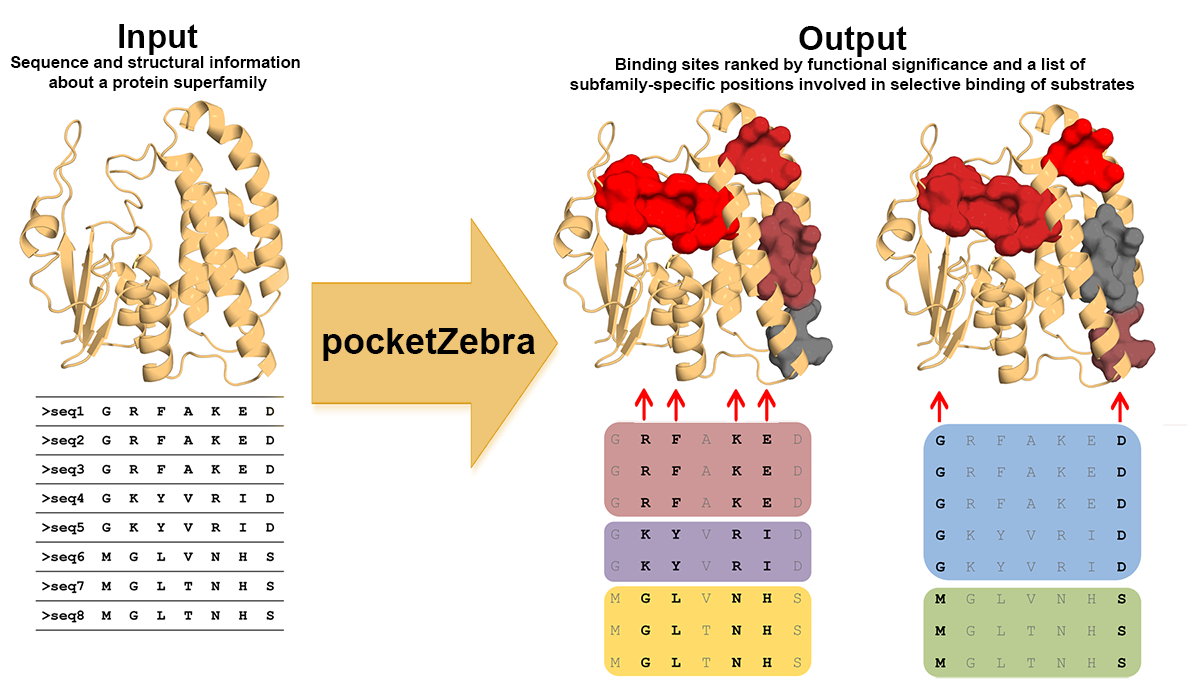
Submission to the pocketZebra should be simple. First, please read our guidelines and the troubleshooting guide concerning the Zebra bioinformatic analysis available at this page. Then, you should further browse the current page for more details about the pocketZebra-specific input. Keep in mind that the input data (first and foremost - the multiple alignment) can be prepared automatically for your particular protein families by the sister web-service Mustguseal using all available information about protein structures and sequences in public databases.
The pocketZebra analysis includes binding sites prediction. If your protein has multiple chains (e.g., A and B) and you multiple alignment represents only a single chain (e.g., A) you should still submit the full-size complete protein structure for adequate prediction of pockets and cavities.
pocketZebra has three input modes
These modes are intended to maximize server usability and accommodate tasks of different complexity.
- pocketZebra in Auto mode - fully-automated-minimum-input. In Auto mode you will need to submit a multiple sequence alignment of a protein family and a representative PDB structure file. Your data will be analyzed with default settings.
- pocketZebra in Pro mode - all-parameters-under-control. In Pro mode you will need to submit the same input data (a multiple sequence alignment of a protein family and a representative PDB structure file) but in addition you will have the full flexibility of fine tuning the algorithm parameters. The advanced knowledge of the pocketZebra procedure is recommended to use this mode.
- Demo mode - Test run demonstration of pocketZebra with pre-selected datasets.
See the Examples page for some real input data.
pocketZebra in Auto mode
You will need to submit a (1) Representative PDB structure file and a (2) Multiple sequence alignment of a protein family. The Multiple sequence alignment should represent a functionally diverse protein family and contain the sequence of the Representative PDB. If the Representative PDB contains multiple chains, you will need to specify the (3) Chain that was used to build the Multiple sequence alignment.

Structural data
- Representative PDB structure:
Upload a PDB file that will be used to identify functionally important pockets and cavities.
See detailsThis structure will be used to identify functionally important pockets and cavities. You should submit a full-size biological unit of your protein because functionally important sites can be located between independent chains. The server automatically corrects your PDB in the following way. First, alternative locations (ex.: ASER/BSER) of the same atom are removed and only the first occurrence of each atom will be retained. Second, Selenomethionine residues (MSE) in the HETATM field will be switched to canonical Methionines under the ATOM field. Therefore, make sure that Methionines in the PDB are also present in the sequence alignment (see Multiple sequence alignment).
Bioinformatic analysis
- Multiple sequence alignment:
Upload a Multiple alignment of a protein family in fasta format.
See detailsThe multiple sequence alignment should represent superimposition of protein amino acid sequences and should be in fasta format. It should contain at least six protein sequences. The latter requirement come from the fact that at least two group (subfamilies) are required to assign the subfamily-specific positions and each group (subfamily) should contain at least three sequences to attain a reasonable level of statistical significance. The "-" sign will be treated as a gap. Standard set of 20 canonical amino acids is allowed. "B", "Z", "X" residue types will be automatically substituted for "D", "E", "G", respectively, with a warning message. Other characters are not allowed.
One of the aligned sequences has to correspond to one of the chains of the Representative PDB structure. In other words the amino acid content of the protein from the Representative PDB file should match one of the protein sequences from the Multiple sequence alignment file with at least 95% pairwise sequence identity. Therefore, to build a valid alignment you should extract the sequence information from the PDB of interest and use it together with other protein sequences of a particular family. You can acquire sequence information about a PDB file at the pdb.org (Display Files >> FASTA Sequence) or by using our command-line Java software PDBParser2.
The algorithm does not require pre-defined classification into subfamilies and can propose multiple classifications automatically by graph based clustering at different fragmentation levels. The proposed functional subfamily classifications are used to identify the subfamily-specific positions. Functional subfamily classifications are finally ranked by significance of SSPs they produce. Alternatively, experimentally derived functional annotation can be provided by the user in the "Pro" mode. - PDB chain:
If the Representative PDB structure contains multiple chains, select the chain that was used to build the Multiple sequence alignment.
pocketZebra in Pro mode
The Pro mode (in contrast to the Auto mode) provides the ability to fine-tune the algorithm parameters that control automatic classification into functional subfamilies, identification of subfamily-specific positions, selection and ranking of functionally important pockets and cavities. Parameters are organized into three separate sections, corresponding to the algorithm workflow:
- Bioinformatic analysis
- Structural analysis
- Statistical analysis
The input data does not differ from the Auto mode. You will need to submit a (1) Representative PDB structure file and a (2) Multiple sequence alignment of a protein family. The Multiple sequence alignment should represent a functionally diverse protein family and contain the sequence of the Representative PDB. If the Representative PDB contains multiple chains, you will need to specify the (3) Chain that was used to build the Multiple sequence alignment.
Structural data
- Representative PDB structure:
Upload a PDB file that will be used to identify pockets and cavities.
See detailsThis structure will be used to identify functionally important pockets and cavities. You should submit a full-size biological unit of your protein because functionally important sites can be located between independent chains. The server automatically corrects your PDB in the following way. First, alternative locations (ex.: ASER/BSER) of the same atom are removed and only the first occurrence of each atom will be retained. Second, Selenomethionine residues (MSE) in the HETATM field will be switched to canonical Methionines under the ATOM field. Therefore, make sure that Methionines in the PDB are also present in the sequence alignment (see Multiple sequence alignment).
Bioinformatic analysis
Run Bioinformatic analysis with default settings
The algorithm parameters are set to their default values.
- Multiple sequence alignment:
Upload a Multiple alignment of a protein family in fasta format.
See detailsThe multiple sequence alignment should represent superimposition of protein amino acid sequences and should be in fasta format. It should contain at least six protein sequences. The latter requirement come from the fact that at least two group (subfamilies) are required to assign the subfamily-specific positions and each group (subfamily) should contain at least three sequences to attain a reasonable level of statistical significance. The "-" sign will be treated as a gap. Standard set of 20 canonical amino acids is allowed. "B", "Z", "X" residue types will be automatically substituted for "D", "E", "G", respectively, with a warning message. Other characters are not allowed.
One of the aligned sequences has to correspond to one of the chains of the Representative PDB structure. In other words the amino acid content of the protein from the Representative PDB file should match one of the protein sequences from the Multiple sequence alignment file with at least 95% pairwise sequence identity. Therefore, to build a valid alignment you should extract the sequence information from the PDB of interest and use it together with other protein sequences of a particular family. You can acquire sequence information about a PDB file at the pdb.org (Display Files >> FASTA Sequence) or by using our command-line software PDBParser2.
The algorithm does not require pre-defined classification into subfamilies and can propose multiple classifications automatically by graph based clustering at different fragmentation levels. The proposed functional subfamily classifications are used to identify the subfamily-specific positions. Functional subfamily classifications are finally ranked by significance of SSPs they produce. Alternatively, experimentally derived functional annotation can be provided by the user in the "Pro" mode. - PDB chain:
If the Representative PDB structure contains multiple chains, select the chain that was used to build the Multiple sequence alignment.
Run Bioinformatic analysis with manual settings
- Multiple sequence alignment:
Upload a Multiple alignment of a protein family in fasta format.
See detailsThe multiple sequence alignment should represent superimposition of protein amino acid sequences and should be in fasta format. It should contain at least six protein sequences. The latter requirement come from the fact that at least two group (subfamilies) are required to assign the subfamily-specific positions and each group (subfamily) should contain at least three sequences to attain a reasonable level of statistical significance. The "-" sign will be treated as a gap. Standard set of 20 canonical amino acids is allowed. "B", "Z", "X" residue types will be automatically substituted for "D", "E", "G", respectively, with a warning message. Other characters are not allowed.
One of the aligned sequences has to correspond to one of the chains of the Representative PDB structure. In other words the amino acid content of the protein in the Representative PDB file should match one of the protein sequences from the Multiple sequence alignment file with at least 95% pairwise sequence identity. Therefore, you are advised to extract the sequence information from the PDB of interest and use it together with other protein sequences of a particular family when creating the final alignment. You can acquire sequence information about a PDB file at the pdb.org (Display Files >> FASTA Sequence) or by using our command-line software PDBParser2.
The algorithm does not require pre-defined classification into subfamilies and can propose multiple classifications automatically by graph based clustering at different fragmentation levels. The proposed functional subfamily classifications are used to identify the subfamily-specific positions. Functional subfamily classifications are finally ranked by significance of SSPs they produce. Alternatively, experimentally derived functional annotation can be provided by the user in the "Pro" mode. - Chain of the Representative PDB structure:
If the Representative PDB structure contains multiple chains, select the chain that was used to build the Multiple sequence alignment. - Gap threshold. Maximal gap occurrence in a column. Columns dominated by gaps usually do not contain any important information.
See detailsExample: set to "30" to remove columns with more than 30% of gaps.
Default: 5% of gaps. - Reference and offset. Select a sequence to be used as reference and an offset value to amino acid position in the sequence in the output file.
See detailsThe two parameters would not affect the calculations.
Example: setting reference to "5" will select the 5th sequence (ex.:ADSST) from the top of the alignment file as a reference. Positions will be shown in the output file as 1A, 2D, 3S, 4S, 5T. Setting offset to "3" would change it to 4A, 5D, 6S, 7S, 8T and could be useful in case the alignment sequence is incomplete and misses first three residues. Offset could be set to a negative value.
Default: 1st sequence is taken as reference with zero offset (positions are numbered according to the order they appear in the sequence - Random permutations. Reliability of statistical calculations is regulated by number of random permutations performed for every columns of the multiple alignment.
See detailsThe default setup is 1000 shuffles per column which provides reasonable precision and fast calculation of your results. This is optimal to get the first impression of how far you can get with your data and also to play with different parameters and setup.
However, when calculating the final draft you may want to set this option to 10'000. This will slow down the process but will provide you with more accurate results.
Example: Setting to "1000" will perform 1000 random permutation in every column of a multiple sequence alignment
Default: 1000 shuffles - Check this box to specify clustering parameters manually. If checked additional parameters are available to manipulate:
- Manual subfamily definition. If pre-defined subfamily classification is available user is welcome to provide it to the program. Classification is submitted as a text file listing space separated sequence ID`s that belong to one family in one line and different subfamilies as different lines (first sequence has id of 1).
Example: Alignment, Groupfile. Perl script can be used to learn sequence id`s from a fasta alignment and assist groupfile preparation. - Predict functional subfamilies. Alternatively, user has an opportunity to process his request in the absence of external functional annotation. Bioinformatic analyses provides a built-in procedure that can be used to predict functional subfamilies.
- Subfamily size limit. Bioinforamtic analyses can create subfamilies with at least 3 sequences (minimally reasonable value).
See detailsProgram was benchmarked with the value of '3' and showed competitive results. However, if you are analyzing a superfamily of thousandths of sequences you are probably not interested in looking at subfamilies of size 3. Thus, to save computational time you can adjust this parameter to the expected size of the smallest subfamily. For CPU time efficiency this parameter has been set by default to 5% of the input sample size but can be changed to any value by the user. If you are not happy with automatically proposed classifications it could mean that functional groups in your alignment substantially vary in size or are too small compared to the overall size of the sample. Try setting "Subfamily size limit" to smaller values (for example: 3 sequences) and repeat the calculation. Example: value of "3" will allow subfamilies with at least 3 sequences
Default: 5% from the number of sequences in alignment but not less then 3 sequences - Outliers . User has an option to select a threshold for outliers (sequences not assigned to a subfamily). Classification exceeding this threshold will be removed.
See detailsSetup for this parameter showed comparable performance in a range 0-30%. Thus, value 20% is set by default. Example: value of '0.2' will make the program to accept classification with not more than 20% outliers compared to the sample size.
Default: 0.2 (not more than 20% from the samle size) - Search by expected number of subfamilies. User has an option to select expected number of subfamilies.
See detailsThis can be set either as a range or as a particular value. Classifications exceeding this threshold will not be considered. Bioinformatic analyses showed significantly better performance when given expected number of subfamilies as an input. Example: setting "mingroups" to "2" and "maxgroups" to "2" will make the program to create only two-group classifications.
Default: number of subfamilies is not limited.
- Subfamily size limit. Bioinforamtic analyses can create subfamilies with at least 3 sequences (minimally reasonable value).
- Manual subfamily definition. If pre-defined subfamily classification is available user is welcome to provide it to the program. Classification is submitted as a text file listing space separated sequence ID`s that belong to one family in one line and different subfamilies as different lines (first sequence has id of 1).
Upload Bioinformatic analysis file
- Upload a raw text output file of the bioinformatic analysis in Zebra format (you can download this file from the results page). In this case, the bioinformatic calculations will be skipped.
Structural analysis
pocketZebra performs the Structural analysis to detect cavities in proteins – potential functionally important sites involved in intermolecular binding. pocketZebra implements the following options at this stage:
- By default, pocketZebra uses the fpocket method. In this respect, if you use the fpocket algorithm to perform the structural analysis please cite:
Schmidtke P., Le Guilloux V., Maupetit J., & Tufféry P. (2010). fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic acids research, 38(suppl 2), W582-W589.
Le Guilloux, V., Schmidtke, P., & Tuffery, P. (2009). Fpocket: an open source platform for ligand pocket detection. BMC bioinformatics, 10:168.
For more information about fpocket please visit http://fpocket.sourceforge.net/.
- Alternatively, you can upload a text file with predefined pockets identified by other algorithms. In this case, please cite the appropriate software.
Run fpocket with default settings
- Default parameters will be used to search for pockets and cavities.
Run fpocket with manual settings
- -m (float): Minimum radius of an alpha-sphere.
Default: 3.0 - -M (float) : Maximum radius of an alpha-sphere.
Default: 6.0 - -A (int) : Minimum number of apolar neighbor for an a-sphere to be considered as apolar.
Default: 3 - -i (int) : Minimum number of a-sphere per pocket.
Default: 30 - -D (float) : Maximum distance for first clustering algorithm.
Default: 1.73 - -s (float) : Maximum distance for single linkage clustering.
Default: 2.5 - -n (integer): Minimum number of neighbor close from each other (not merged otherwise).
Default: 3 - -r (float) : Maximum distance between two pockets barycenter (merged otherwise).
Default: 4.5 - -p (float) : Minimum proportion of apolar sphere in a pocket (remove otherwise).
Default: 0.0 - -v (integer): Number of Monte-Carlo iteration for the calculation of each pocket volume.
Default: 2500
Manual definition of pockets
Upload a file that defines pockets in your structure. In this case, the structural analysis will be skipped. The file should state the amino acids that are involved in formation of structural sites. This information can be obtained by any third-party pocket-searching algorithm or hypothesized by the user. In both cases pocket definition should be translated to the format that can be understood by pocketZebra.
See details
- Each line contains space-separated keywords that define a new pocket
- Each line starts with pocket identifier - POC[XX], that contains letters POC followed by a non-negative integer [XX]
- Every other keyword in a line represents a particular atom of an amino acid that is involved in pocket formation. Two formats are available:
- [CHAIN]/[RESID]/[RESNAME]/[ATOM], CHAIN and RESNAME represent chain identifier and residue name, respectively, both written in letters (both upper and lower cases are accepted); RESID - non-negative integer that represents residue ID; ATOM - a combination of letters (both upper and lower cases are accepted) and numbers that represent the atom name. This combination should be unique (appear only once) within the PDB file.
Example:
POC0 A/216/ASP/N A/216/ASP/CB A/55/LYS/NZ A/55/LYS/CE A/216/ASP/CA A/216/ASP/OD1 A/216/ASP/CG A/216/ASP/OD2 A/55/LYS/CD A/55/LYS/CG
POC1 A/64/ASP/OD2 A/64/ASP/OD1 A/155/ARG/NH2 A/155/ARG/CD
POC2 A/268/ASP/OD1 A/219/ARG/NE A/148/ARG/NH2 - [CHAIN]/[RESID]/[RESNAME]/-, all atoms of a particular amino acids will be automatically retrieved from the PDB file and included in the corresponding pocket.
Example:
POC0 A/216/ASP/- A/55/LYS/-
POC1 A/64/ASP/- A/155/ARG/-
POC2 A/268/ASP/- A/219/ARG/- A/148/ARG/- - a pocket can be defined by a combination of keywords using the two different formats.
Example:
POC0 A/216/ASP/- A/55/LYS/-
POC1 A/64/ASP/- A/155/ARG/-
POC2 A/268/ASP/OD1 A/219/ARG/NE A/148/ARG/NH2 - pocketZebra ranks the binding sites by specificity of corresponding amino acid residues and does not use information about particular atoms. Therefore, the format in which a residue is defined (by specifying particular atoms or the whole residue) will not affect the calculations but will change the visual appearance of the corresponding pocket in the results.
- empty lines will be skipped
- duplicate keywords are not allowed
- situations when duplicate atoms appear in the same pocket are not allowed. For example, the following line in the pocket definition file will throw an error because the backbone nitrogen will appear twice in POC0 - first from A/216/ASP/- and then explicitly from A/216/ASP/N.
Example:
POC0 A/216/ASP/- A/216/ASP/N - empty pocket definitions (that do not contain any residues) are not allowed
- [CHAIN]/[RESID]/[RESNAME]/[ATOM], CHAIN and RESNAME represent chain identifier and residue name, respectively, both written in letters (both upper and lower cases are accepted); RESID - non-negative integer that represents residue ID; ATOM - a combination of letters (both upper and lower cases are accepted) and numbers that represent the atom name. This combination should be unique (appear only once) within the PDB file.
- The pocket definition file should be in plain text format and have a .txt extension.
- Download an example pocket definition file that corresponds to Example #3 from the Examples page.
Statistical analysis
This section provides the ability to control the selection and ranking of binding sites by specifying the cut-off method to choose significant subfamily-specific positions. Two modes are available at the input:
- By global P-value minimum (default). A set of subfamily-specific position with the smallest P-value will be selected as the most significant.
- By Z-score cut-off. Define the smallest (inclusive) Z-score for a subfamily-specific position to be considered when selecting and ranking the functionally important sites.