Mustguseal Parameters
The user can manipulate the key parameters of the two crucial steps in the Protocol - the Structure similarity search and the Sequence similarity search. These parameters define the scope of the final structure-guided sequence alignment, i.e., how evolutionary distant, structurally and functionally diverse are the proteins used for the superimposition. This pages gives an overview of these parameters and explains their influence over the results. The detailed guidelines for parameters selection and examples are provided in the Supplementary Data to Mustguseal publication.
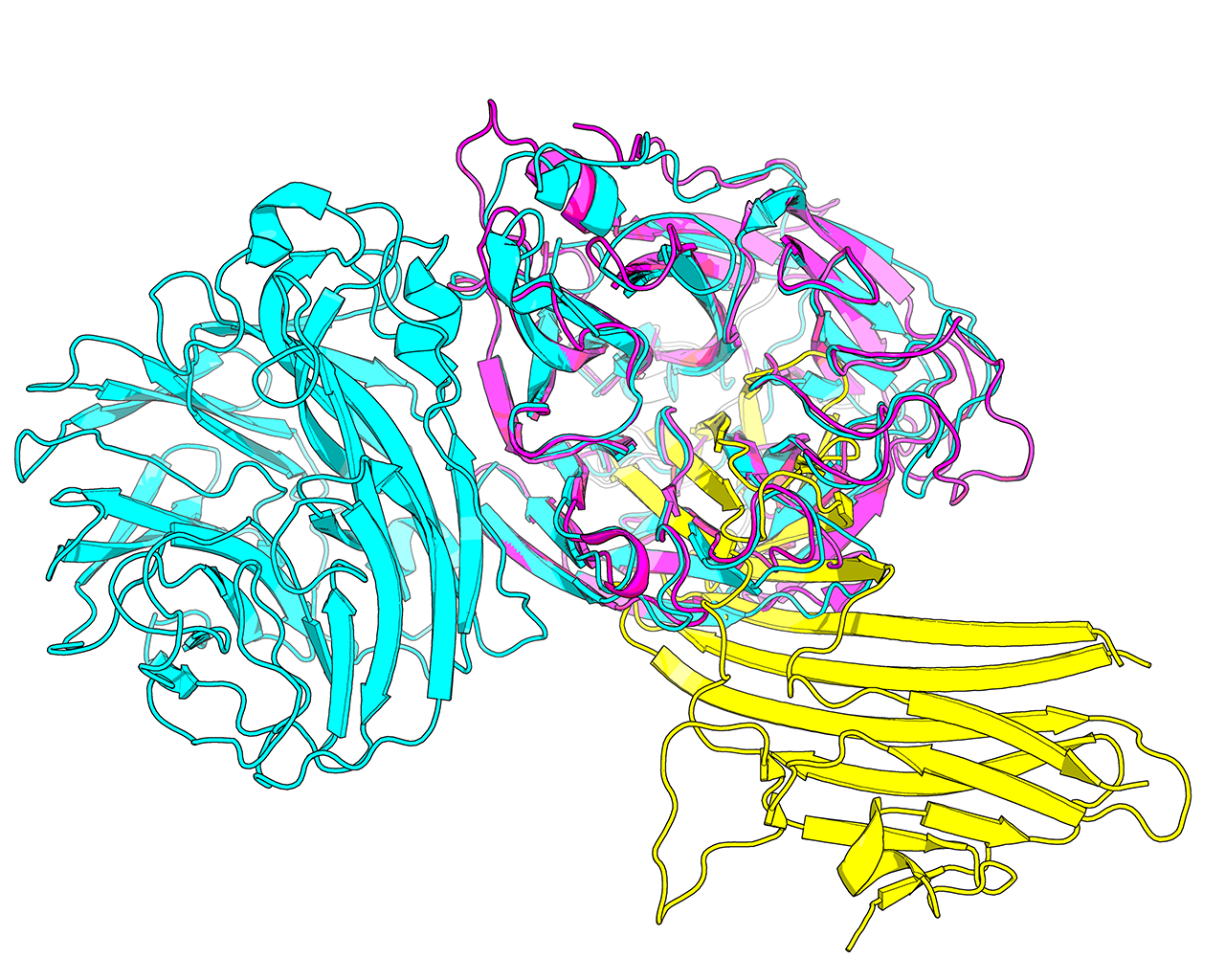
Protein structure is considered to be more conserved throughout the evolution compared to sequence. Therefore, structure similarity search versus the entire PDB database is used by Mustguseal to collect remote evolutionary relatives of the query protein by selecting hits with a sufficient similarity in structural organization. In Mustguseal the structural similarity is quantified by the alignment coverage - i.e., the ratio of the number of amino acids in the aligned (superimposed) parts of the query and the target structures compared to the full size of the corresponding structures. Only amino acids involved in secondary structure elements are considered to calculate the ratio. By default the thresholds are set to 90% and 90%, i.e., at least 90% of the query have to make at least 90% of the target for it to be selected as a hit. The choice of these thresholds should be individual for each task and should take into account the structural characteristics of a particular family. E.g., the example above shows a single-chain single-domain query protein (magenta) being matched with a single-chain dual-domain target protein (cyan). Approximately 90% of the query makes approximately 50% of the target (i.e., the full query structure is involved in superimposition with the target, but at most half of the target structure is involved in superimposition with the query). To select this target as a match with the query the thresholds should be set to approximately 90% (of the query structure) and 45% (of the target structure).
Generally speaking, if the aim is to construct a more diverse alignment, or if no structures or too few structures similar to the query were found by the structure similarity search with the default setup, the user should consider decreasing the Lowest acceptable match in the query and target structures in the Structure similarity search input box to 70% and 70%, respectively. Proteins which are more evolutionary remote will be collected increasing structural and functional variability of the alignment. The 70%-70% setting can be used to obtain a diverse set of functions within a common structural framework, given equivalent structural dimensions of the proteins in a superfamily. If the dimensions of protein structures (i.e., domain organisation) are not equivalent among the protein families of interest (e.g., see the magenta-cyan case above), one of the parameters should be set to 70% and the other one should be set below 70%, but not less than 30%.
Please note, that at least some level of similarity may be found in almost any pair of protein structures picked at random from the PDB database. The aim of the structure similarity search is to collect structurally and, thus, functionally diverse but yet evolutionary related proteins to serve as the core for the structure-guided sequence alignment. Homologous proteins within a superfamily may have significantly diverged in both sequence and structure during the evolution. Therefore, decreasing the two thresholds to a very low values (e.g. 30% and 30%) may help in identifying all available members of a superfamily of interest, but significantly increases the probability of collecting unrelated (i.e., not homologous) proteins and corrupting the alignment. E.g., the example above shows a 30%-30% match between the query protein (magenta) and the target protein (yellow). It is clear from this comparison that the two proteins are unrelated and their alignment would be meaningless in the context of Mustguseal protocol.
The user can also choose whether to consider the entire PDB database or only the structures obtained using the X-ray crystallographic analysis for the structure similarity search. If the input query protein is not an X-ray structure then the The entire PDB database option should be checked, otherwise the task will be rejected.
Regarding the Select at most representative proteins parameter. This option makes sense when running in the Mode 1 with UniProtKB/Swiss-Prot+TrEMBL databases or in the special-purpose Mode 4. A brief description of the special-purpose Mode 4 can be found here. Regarding the Mode 1, this parameter can regulate the maximum number of protein 3D-structure that will be automatically selected for the non-redundant representative set. Historically, this parameter was hard-set to at most 32 when subsequently using the UniProtKB/Swiss-Prot database for sequence similarity searches, and to at most 16 when additionally using the significantly larger TrEMBL database. Since June, 2020 the user can choose between 16 and 32, for a particular purpose. We urge the users to be reasonable, e.g., adding another 16 representative proteins to your core 3D-structural alignment can add another ~16'000 protein sequences to your final alignment when using TrEMBL (i.e., if the "Maximum number of sequences to collect in each subsearch" parameter was set to "1000"). Larger alignments indeed contain more data, but it is only useful if you know how to analyze/process such large alignments. In most cases, a smaller alignment is easier to study and therefore more likely to contribute to your research compared to unreasonably large alignment. More details about the Mustguseal performance and limitations are provided here.
Regarding the Select representative set below 40% sequence identity parameter. By default, all protein structures collected by the structure similarity search are clustered by similarity of the corresponding sequences at 95%, 90%, ..., 40% identity thresholds to select at most 32 representative proteins for further superimposition and processing (see "The Protocol" section in the Supplementary Data file to the Mustguseal paper and the Mustguseal Performance page). If the number of unique proteins exceeds 32 after clustering at the 40% threshold then only 32 proteins which are the most structurally similar to the query are selected automatically and the remaining structures are dismissed. By setting this option to the "Yes" value the user can instruct Mustguseal engine to continue with selecting the representative set of protein structures below the 40% sequence similarity threshold (down to 10% with a 5% step). Setting this option to "Yes" can help to collect the most diverse set of proteins for a better representation of a large superfamily, however can result in a selection of proteins which are very distantly related to each other. In order to compensate for the diversity of the representative proteins set and collect information about the intermediate proteins it is advised to alter filter settings in the "Sequence similarity search" section: set the "Maximum number of sequences to collect in each subsearch" to 1000, the "Redundancy filter threshold" to 80%, and the "Dissimilarity filter threshold" to 0.25 (or below, although using the lower values of this parameter could prompt errors by allowing too distant proteins in the sequence alignment). An alternative to using the "Yes" option to build the alignment of a very large superfamily would be to use Mode 1 to run the structure similarity search, then download the results and pick the representative set manually, and then use Mode 2 (or a combination of Modes 2 and 3, depending on the size of the representative set) to construct your alignment. See the Mustguseal Parameters for a guideline to construct a custom core structural alignment for Mode 2.
Sequence similarity search
Each representative protein from the core structural alignment is used as a query to run a sequence similarity search. The following parameters influence the outcome of this step.
Selection of a database. The use of UniProtKB/Swiss-Prot database is set as the default. This database provides protein sequences as well as, in general, a trustworthy functional annotation. The downside is its relatively small size. Not all protein families are fairly represented in the Swiss-Prot database. The user could try the UniProtKB/Swiss-Prot+TrEMBL database which is much larger and usually provides more proteins for the alignment. The downside is that the functional annotation provided by the TrEMBL is a prediction (i.e., annotation transfer by similarity with well-studied proteins) and should be considered for information purposes only.
Maximum number of sequences to collect in each subsearch. If the UniProtKB/Swiss-Prot database was selected then three subsearches would be carried out - using the Blosum45, Blosum62, and Blosum80 matrixes. If the UniProtKB/Swiss-Prot+TrEMBL database was selected then four subsearches would be carried out - three in the Swiss-Prot database using the Blosum45, Blosum62, and Blosum80 matrixes, and one in the TrEMBL database using the Blosum62 matrix. The Maximum number of sequences to collect in each subsearch parameter limits the maximum number of sequences to be collected after each subsearch. Keep in mind, that this parameter regulates only the size of the 'raw' set, and all sequences collected by the similarity search will be further filtered for sequence length, redundancy* and dissimilarity with the representative protein (see description of the three filters below, in particular you should see the (*) description in the redundancy filter section), thus the size of the final set of sequences can be significantly reduced. The default value of 500 for this parameter can be implemented for good in the absolute majority of cases. If your final alignment turned out to be too large you may want to decrease this parameter (e.g., to 50) to limit the number of proteins collected during the similarity search. If no sequences or too few sequences similar to the representative protein were found by the sequence similarity search you should check the output logs for the corresponding search (i.e., the seqsearch_PDBID.stdout.log file in the BLAST_PDBID folder, see the Explanation of the Output for more details). If the number of proteins collected in each subsearch is up to the limit (e.g., 500 proteins) but they are being dismissed as redundant (too similar to each other) during the further filtering, you could increase this parameter (e.g., to 1000) in an attempt to collect more diverse proteins.
Redundancy filter threshold (%). Sequences collected by sequence similarity search in the selected database are further filtered for non-redundancy. By default, only one sequence is preserved from each cluster of sequences which share at least 95% similarity, and all others are dismissed from further consideration. Aligning sequences with more than 95% identity would increase the computation cost while adding doubtful information value. Therefore, releasing this threshold (i.e., setting it to values >95%) could be justified only for a particular purpose. The user could consider tightening this threshold (i.e., setting it to values <95%) to reduce the total number of proteins when constructing alignments of very large superfamilies (e.g., the alpha-beta hydrolases). Generally speaking, you should not aim at constructing a very large alignment (>5000-10000 proteins). Very large alignments are impractical as they would most certainly contain redundant information and would be computationally hard to analyze.
(*) Please note that the pre-calculated non-redundant sets of the UniProtKB/Swiss-Prot and UniProtKB/TrEMBL databases are actually being used by the server to accelerate the sequence similarity searches - the databases are clustered at the 100% (nr100), 95% (nr95) and 80% (nr80) sequence identity thresholds:
- if the user-defined Redundancy filter threshold is set to (95-100%], then the nr100 set of sequences is used;
- if the user-defined Redundancy filter threshold is set to (80-95%], then the nr95 set of sequences is used;
- if the user-defined Redundancy filter threshold is 80% or below, then the nr80 set of sequences is used.
E.g., if the user-defined Redundancy filter threshold is set to 90%, then the nr95 sequence set derived from the UniProtKB/Swiss-Prot or UniProtKB/TrEMBL is used to run a series of sequence similarity searches, and the collected sequences are further filtered for non-redundancy at the 90% sequence similarity threshold. The size of the sets, and, consequently, the time required to perform a sequence similarity search in the corresponding sets, differ as follows: nr80 < nr95 < nr100. Consequently, the value of the Redundancy filter threshold has a direct impact on the speed of the sequence similarity searches (a value below 80% is the fastest option, 100% is the slowest option).
Dissimilarity filter threshold (bit score per column). The selected set of proteins is further filtered to eliminate too distant proteins which would likely cause errors during the sequence alignment. The dissimilarity threshold is quantified in bit scores per column and describes the entropy per column in the pairwise alignment of a selected protein with the representative protein (which was used as a query to run this similarity search). By default, proteins with at least 0.5 bit score per column with the representative protein are preserved, and all others are dismissed from further consideration. Set this parameter to a higher value to preserve only proteins which are more similar to the query, or to a lower value to allow less similar proteins in the sequence alignment. The bit score per column may take a wide range of values, however, the most commonly used are values within the [0; 1] range (usually 0.25 or 0.5). Therefore, not to confuse the user with a large selection of rarely used settings, we limit the Dissimilarity filter threshold to [0; 1].
Sequence length filter threshold (%). By default, proteins which differ by more than 20% in length from the representative protein used as query to run the sequence similarity search are removed to eliminate too small and too large protein sequences which can correspond to incomplete or incorrect database entries, and thus to decrease the number of columns which are highly gapped. This Sequence length filter should be released if your representative proteins correspond to fragments of protein chains or proteins which are represented by a larger precursor sequences in the sequence database. If you get a similar warning
Warning: Sequence similarity search for the representative protein 0_1gm9_B has returned only itself
you should download the archive with sequence similarity search results, enter the corresponding folder (i.e, BLAST_0_1gm9_B for this example), and check the log (seqsearch_0_1gm9_B.stdout.log) for output like this:
Info: Sequence P06875 has length 846 and will be dismissed (151.9 % of the reference sequence length)
Info: Sequence P07941 has length 844 and will be dismissed (151.5 % of the reference sequence length)
Info: Sequence P15558 has length 774 and will be dismissed (139.0 % of the reference sequence length)
This warning happens because the P06875, P07941, and P15558 are sequences of the precursor protein, which includes chain A, the linker, and chain B, while the PDB file 1GM9:B corresponds to the chain B only. In order to include sequences of proteins P06875, P07941, and P15558 in the alignment set the Sequence length filter threshold to 60% (i.e., to allow variations in length in a range 40%-160% of the length of reference protein sequence), or you may effectively switch the filter off by setting it to 9999%.
"Use MAPU to build large alignments" parameter. Setting this parameter to "Yes" will enable trimming of the alignment by the sequences of representative proteins, i.e., the alignment columns which contain a gap in all representative proteins will be removed. The output final alignment will contain the complete sequences of the representative proteins aligned with each other and subsequences of their homologues. The trimming will not lead to loss of information because the analysis of the final alignment by the sister web-servers of Mustguseal is focused only on the columns which contain amino acid residues of the representative proteins, and this information will be identical in the original and trimmed versions of the alignment. The purpose of trimming is to decrease the size of the final alignment file which can be crucial when dealing with large sets of diverse sequences and structures. E.g., the untrimmed version of the final alignment may occupy several hundred megabytes of the disk space due to a high content of gaps, and trimming can decrease the size by ~98% percent to just a few megabytes, which is very convenient for further analysis.

We have developed the Mustguseal Alignment Postprocess Utility (MAPU) to perform the trimming and thus to reduce the disc space and RAM required to store and process a multiple alignment of a diverse set of evolutionary distantly related homologues by preserving the most informative columns and removing the least informative once. The MAPU modifies the alignment by removing columns which contain a gap in all representative proteins, thus making it more compact, portable, and convenient for further visual and automatic analysis. The output final alignment will contain the complete sequences of the representative proteins aligned with each other and subsequences of their homologues. Such a trimmed version of the alignment, while retaining the most content-rich columns, is usually much smaller in size and thus requires a significantly less computational effort at further processing. The use of MAPU at alignment construction significantly decreases the computation requirements, and thus provides an opportunity to construct a very large multiple alignment. When MAPU is enabled the following limits are raised:
- Maximum number of sequences to collect in each subsearch is raised to 5000 proteins in Modes 1 and 2;
- The maximum size of the final alignment in Mode 3 is raised to 50 000 proteins.
Please note, that current web-based implementations of sister web-servers of Mustguseal have the following limitation on the size of the input multiple alignment: Zebra - at most 15000 proteins, pocketZebra - at most 15000 proteins, visualCMAT - at most 50000 proteins, and Yosshi - at most 50000 proteins.