Dealing with the multiple-chain proteins
The Yosshi analysis is provided on a one-chain-at-a-time basis. If your protein contains multiple chains and you are interested in the analysis of disulfides which crosslink different chains (i.e., inter-chain/inter-subunit S-S bonds) you need a workaround. The quick guide below provides an easy-to-use solution.
- The workaround for multiple-chain proteins
- Overview of the software
- Comments on dealing with homo-polymers
- Comments on dealing with hetero-polymers
The workaround for multiple-chain proteins
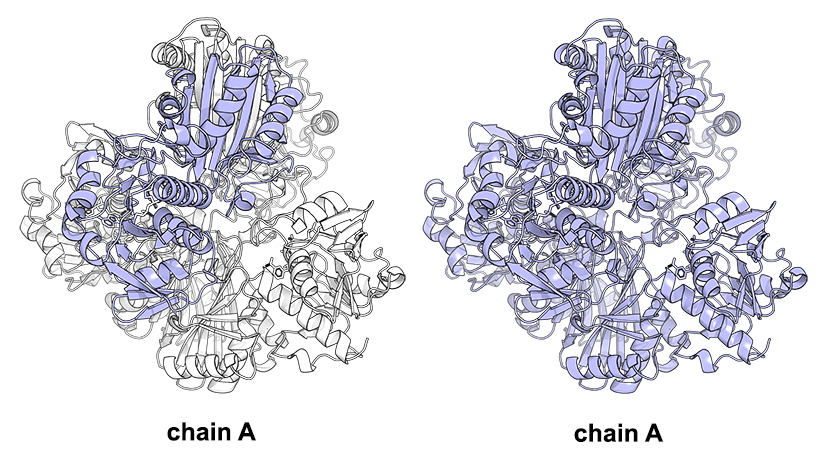
As discussed in the respective section the Yosshi web-server requires (1) a multiple alignment and (2) the query protein structure for input. You need to see that page for the general guidelines to prepare the input data. The Yosshi then operates on a single-chain basis. I.e., the web-server takes the particular user-defined protein chain from the PDB file and matches it with the alignment for further processing. If the query protein contains multiple chains this approach will detect only the intra-subunit disulfides (i.e., crosslinks between Cysteines within one chain) and will not detect the inter-subunit S-S bonds. Although the inter-subunit disulfides, in general, present a relatively rare event, they were proven by several studies to play a key role in the protein structure and considered useful at protein engineering. The workaround for detecting the inter-subunit S-S bonds using Yosshi would be to present a multiple-chain protein as a single-chain one. The chain identifiers of multiple chains in the PDB file can be changed to a single code (e.g., chains A, B, C, and D are renamed to A) followed by renumbering of residue IDs, to produce a single-chain PDB file for further analysis by Yosshi. I.e., the amino acid residues, their names, coordinates and sequence are preserved in full, but the chain IDs and residue numbering is fixed for compatibility with Yosshi - see the figure above. The same applies to the sequence alignment - multiple alignment files are merged into one, in accordance with the order of chains in the PDB file. The details are provided below.
Overview of the software
The Software: Multiprot.pl v. 1.1 [download]
Download the script and use it to merge PDB and alignment data for your multiple-chain protein. The software operates in two modes.
Use the pdb mode to process the structural data:
>./multiprot.pl pdb [input.pdb] [chainID] [chainID] [optional=chainID] [optional=chainID] ... [output.pdb]
[input.pdb] - Path to the input PDB structure of a multichain protein
[chainID] - Provide at least two IDs of protein chains to merge
[output.pdb] - Path to the write the output PDB structure after processing
Use the aln mode to process the sequence alignment data:
./multiprot.pl aln [input.fasta] [input.fasta] [optinal=input.fasta] [optinal=input.fasta] ... [output.fasta]
[input.fasta] - Provide at least two alignment files corresponding to different protein chains to merge
[output.fasta] - Path to the write the output alignment after processing
The output of the pdb mode is a PDB file with renamed chains and renumbered amino acid residues, and a multiprot_pdb.log file with the correspondence table between the initial and output residue numbering. The output of the aln mode is a FASTA multiple alignment. The produced FASTA and PDB files can be submitted to Yosshi for the analysis.
Comments on dealing with homo-polymers
If your protein is a homo-polymer consisting of identical chains then its processing would be straightforward. First, use the pdb mode of the multiprot.pl script (the download link is in the section above) to rename all chains of the PDB file to appear as a single chain, then use the aln mode to create a copy of the alignment for each chain and merge them together into a single alignment file. The examples below perform these operations for a homotetermetic GAPHD protein (PDB: 1GD1) which contains chains A, B, C, and D.
Processing of the PDB structure file of a homotetrametic protein (i.e., chains A, B, C, and D of the PDB file are renamed to chain A)
>./multiprot.pl pdb 1gd1_mm1.pdb A B C D 1gd1_ABCD.pdb
Input: INPUT PDB 1gd1_mm1.pdb
Input: INPUT CHAINS [A B C D]
Input: OUTPUT PDB 1gd1_ABCD.pdb
Input: OUTPUT CHAIN_ID A
Info: Reading the input PDB file 1gd1_mm1.pdb
Info: Renaming chains [A B C D] to chain A and updating the numbering
Info: Found 2754 atoms in chain A -> converted to chain A
Info: Found 2740 atoms in chain B -> converted to chain A
Info: Found 2744 atoms in chain C -> converted to chain A
Info: Found 2742 atoms in chain D -> converted to chain A
Info: Writing the processed PDB to 1gd1_ABCD.pdb
Info: The correspondence table between the input and output residue numbering has been printed to multiprot_pdb.log
Done!
Processing of the FASTA alignment file of a homotetrametic protein (i.e., the alignment file chainA.fasta of the chain A is copied four time and merged into a single alignment file)
./multiprot.pl aln chainA.fasta chainA.fasta chainA.fasta chainA.fasta chainsABCD.fasta
Input: INPUT FILE SEQUENCE CONTAINS 4 ENTRIES
Input: INPUT ALIGNMENT #1 chainA.fasta ! (used 4 times)
Input: INPUT ALIGNMENT #2 chainA.fasta ! (used 4 times)
Input: INPUT ALIGNMENT #3 chainA.fasta ! (used 4 times)
Input: INPUT ALIGNMENT #4 chainA.fasta ! (used 4 times)
Input: OUTPUT ALIGNMENT chainsABCD.fasta
Info: Reading the input multiple alignment files
Info: Alignment file chainA.fasta contains 16670 sequences and 1156 columns
Info: Looking for common sequences which are present in all input alignment files
Info: All input alignment files contain a common list of 16670 sequences
Info: Merging alignments of common sequences in the requested order and printing the final alignment to chainsABCD.fasta
Info: The merged alignment contains 16670 sequences and 4624 columns
Done!
These operations will produce three output files. The produced files 1gd1_ABCD.pdb and chainsABCD.fasta can be submitted to Yosshi for the analysis. The file multiprot_pdb.log contains the correspondence table between the residue numbering in the initial PDB file 1gd1_mm1.pdb and the processed PDB file 1gd1_ABCD.pdb.
Comments on dealing with hetero-polymers
The procedure is qualitatively the same for hetero-polymer proteins containing different subunits. The different chains in the PDB file are renamed and renumbered to appear as a single chain, and the alignment files are merged. However, when merging alignment files corresponding to different chains only those sequences are preserved which belong to the same protein defined by identical name in all files. I.e., if two files were provided for input - chainA.fasta and chainB.fasta - then only those sequences within these files that have the same name (meaning that they are part of the same protein) will be merged together, and the remaining sequences (which are parts of different proteins) will be dismissed from further consideration. This has to be taken into account when constructing the respective multiple alignments. Example:
./multiprot.pl aln chainA.fasta chainB.fasta chainC.fasta chainsABC.fasta
Input: INPUT FILE SEQUENCE CONTAINS 3 ENTRIES
Input: INPUT ALIGNMENT #1 chainA.fasta
Input: INPUT ALIGNMENT #2 chainB.fasta
Input: INPUT ALIGNMENT #3 chainC.fasta
Input: OUTPUT ALIGNMENT chainsABC.fasta
Info: Reading the input multiple alignment files
Info: Alignment file chainA.fasta contains 16455 sequences and 1156 columns
Info: Alignment file chainB.fasta contains 6326 sequences and 1156 columns
Info: Alignment file chainC.fasta contains 16670 sequences and 1156 columns
Info: Looking for common sequences which are present in all input alignment files
Info: There are 6111 sequences which are present in all input alignment files
Info: Cleaning alignment data from sequences which are not present in all input alignment files
Info: Cleaning the alignment chainA.fasta
Info: Removed 767 gaps-only columns in chainA.fasta after clean-up
Info: Cleaning the alignment chainB.fasta
Info: Removed 767 gaps-only columns in chainB.fasta after clean-up
Info: Cleaning the alignment chainC.fasta
Info: Removed 767 gaps-only columns in chainC.fasta after clean-up
Info: Merging alignments of common sequences in the requested order and printing the final alignment to chainsABC.fasta
Info: The merged alignment contains 6111 sequences and 1167 columns
Done!