Yosshi parameters
The Yosshi workflow consists of bioinformatic analysis to search for pairs of Cysteine residues in sequences of homologs, and structural filtration to evaluate whether introduction of the selected Cysteines at corresponding positions of the user-submitted representative (query) protein structure can result in a formation of a disulfide bond. This section describes the corresponding parameters of the Yosshi analysis available to the user and their influence over the output.
- First tier filter
- Bioinformatic analysis
- 3D-motif analysis
- Thresholds for structural filtration
- Provide HTML links to PDB, UniProt, and BacDive
The following key parameters define the Yosshi process and can be edited by the user:
- The dist(Cα−Cα) and dist(Cβ−Cβ) to define the initial set of candidate pairs of positions in the query protein structure;
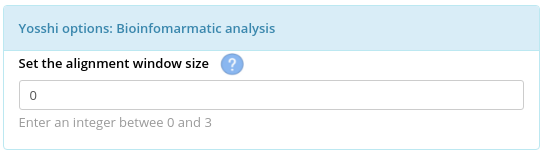
- The alignment window size to manage how sequences of homologs are mapped onto the query protein structure;
- The parameters for 3D-motif analysis: the choice of a 3D-motif library and parameters of a statistical model for 3D-motif comparison with the query protein - mean μ, standard deviation σ, and a p-value of a normal distribution;
The details are provided below.

The “First tier filter” represents one of the two structural filtration steps and is applied to the query protein structure to dismiss all pairs of positions which are the least likely to form a crosslink even if both were mutated to Cysteines using simple dist(Cα-Cα) and dist(Cβ-Cβ) distance criteria. This step is casually performed by disulfide engineering algorithms, but the threshold selection is usually based on the restrictive model of two covalently bound Cysteines, e.g., all amino acid pairs are considered as potential hot-spots if their Cβ atoms are within 4.5 Å, or 5.21 Å, or 5.5 Å. There are numerous reports of successfully engineered disulfide bonds that violate geometric constraints of these rigid computational models. Thus, in this web-server a more flexible geometry model is applied – the cut-off values are set to dist(Cα−Cα)≤8.58 Å and dist(Cβ−Cβ)≤6.96 Å, which correspond to μ+3σ of the corresponding values calculated over the set of 123 non-bonded equivalences E1-E2 of disulfide bonds in homologous proteins (see below). The aim of the “First tier filter” is to accelerate further processing by removing the least appropriate candidate positions. Therefore, a low first-order error of 0.135% (i.e., P(x>μ+3σ)=0.00135 assuming the distances are normally distributed) is considered for threshold selection to minimize the risk of rejecting two candidate positions which may it fact form a correct disulfide bond, at a cost of failing to reject two candidate positions which are incapable of a disulfide bond formation, as this initial selection is further evaluated by two more steps of the algorithm, in particular, a more detailed analysis of the geometry of each two positions is performed at the last step of the workflow by the 3D-motif analysis (see protocol for an overview of the Yosshi workflow).
Yosshi searches for pairs of Cysteine residues in sequences and structure of homologs and evaluates whether introduction of these Cysteines at corresponding positions of the representative (query) protein structure can result in a formation of a disulfide bond. The bioinformatic analysis is a key part of Yosshi protocol and requires that a large set of proteins which are expected to have diverse properties and different disulfide connectivity patterns within a common structural core of a superfamily is collected and aligned, thus representing a methodological challenge. This step is facilitated by the integrated Mustguseal web-server capable of automatically constructing large structure-guided sequence alignments of functionally diverse protein families, as described here. Comparison of the collected homologs with the query protein is carried out according to the alignment, i.e., by default, Cysteine residues in sequences of homologs are mapped onto those positions in the query protein structure which are situated in the same columns of the multiple alignment. A more broad interpretation of the multiple alignment can be performed by changing the "Alignment window size" parameter.
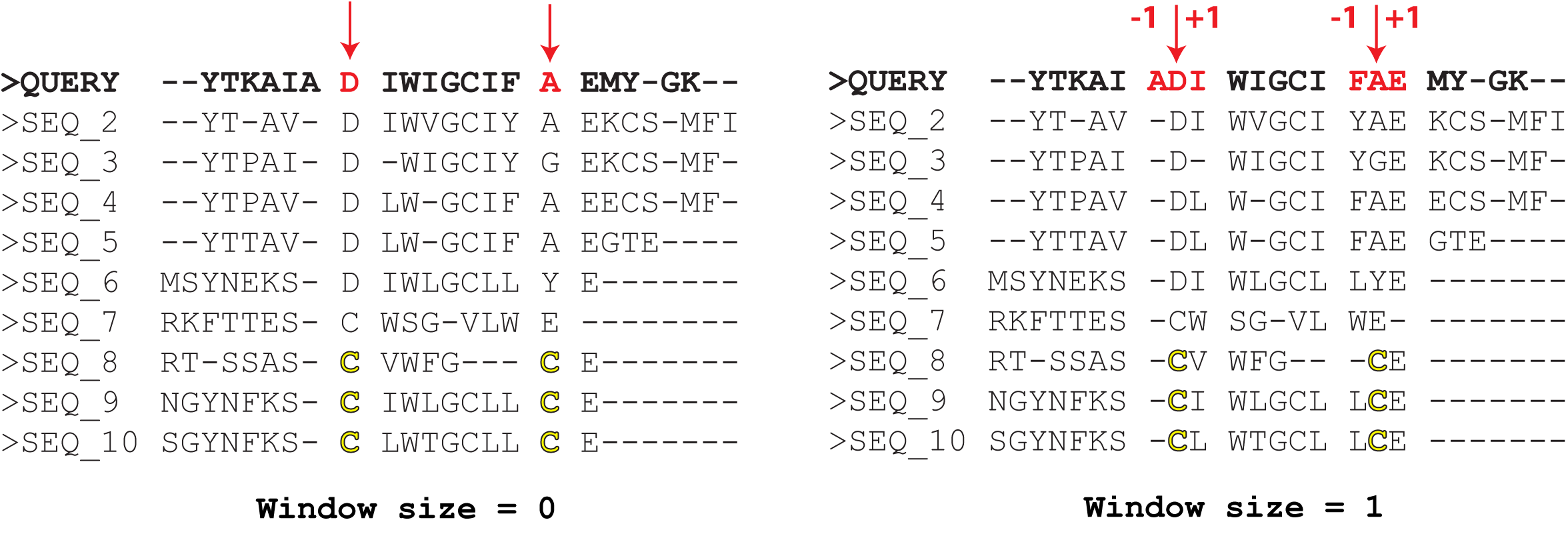
The "Alignment window size parameter defines how the Cysteine residues in sequences of homologs are mapped onto the query protein sequence/structure. By default, this parameter is set to zero, meaning that the query protein will be matched with other homologs exactly according to the alignment - i.e., a Cysteine residue in the alignment column i in a homolog will be mapped onto residue X of the query protein in the very same alignment column i (see the left figure above). Setting this parameter to, e.g., "1" would consider three alternative candidate positions to map Cysteine residues of a homolog in the alignment columns i and j onto the query sequence - at the alignment columns i-1, i, i+1, and j-1, j, j+1, respectively (see the right figure above). Then, the corresponding 3 x 3 = 9 combinations of every two positions will be evaluated by the 3D-motif analysis to select only those positions in the query protein structure that satisfy the geometric constraints of the disulfide bond formation. The key idea of this approach is that protein sequence/structure comparison algorithms can make mistakes, and as a result amino acid residues of sequences/structures of homologs can be misplaced/shifted in the alignment. Setting the "Alignment window size" to a non-zero value can help to correct these minor alignment errors by considering alternative interpretations of how to match the sequences of homologs. The catch is, this convenience may increase the False Positive Rate, i.e., it may produce too many predictions and not all of them will be true.
To conclude, setting the "Alignment window size" parameter to a non-zero value should NOT be used as a global measure to predict disulfides in the full-size protein, but rather as a local one to refine the predictions in a selected regions. E.g., if you expect a disulfide bond to be formed between two particular regions in your query structure (e.g., two loops), but Yosshi fails to produce a result with the default parameters, you could activate this feature and then evaluate the predictions in these particular regions manually.
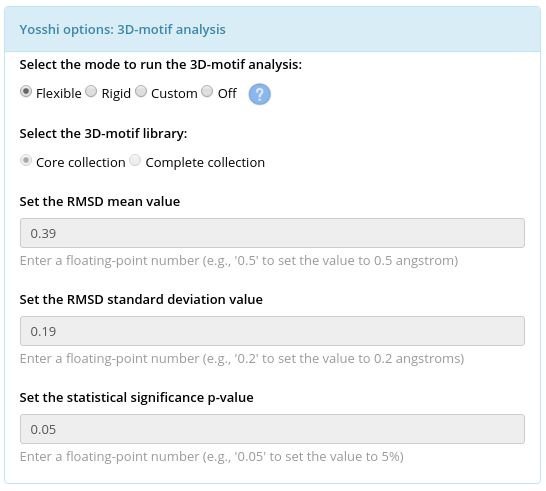
The 3D-motif analysis is the last step of the Yosshi protocol used to evaluate whether each pair of positions selected by the first tier filter and the bioinformatic analysis in the representative protein structure (i.e., both positions are occupied by Cysteines in at least one homolog) can form a disulfide bond assuming the respective amino acids are mutated to Cysteines in the representative protein, and prepare 3D-models of the corresponding mutants. When the "Rigid" mode is used for the 3D-motif analysis only those positions in the query protein structure will be approved as promising sites for S-S bond engineering whose geometry is highly similar to the geometry of the known covalently connected Cysteine residues. On the contrary, when the "Flexible" mode is used for the 3D-motif analysis the backbone flexibility will be taken into account to approve a pair of positions that does not match strict geometric constraints of an S-S bond formation in the provided "static" crystallographic structure but may still form a crosslink when both mutated to Cysteines due to a shift of the backbone atoms. If the 3D-motif analysis is turned OFF then this last step will be skipped, and all pairs of positions selected by the first tier filter and the bioinformatic analysis will go straight to the output, i.e., the output will list all disulfides in homologs mapped onto the representative protein structure without evaluating if these disulfides are actually capable to form an S-S bond. By default, the 3D-motif analysis is turned ON in the "Flexible" mode. The details are provided below.
In the context of this study, a 3D-motif of a disulfide bond contains 12 atoms of the backbone and side-chain of two covalently linked Cysteines taken from a high-quality crystallographic structure (see an example below). Two collections of 3D-motifs were created based on the analysis of the PDB database and incorporate the currently available knowledge of the disulfide bond geometry: the “Core collection” featuring 273 most typical configurations of S-S bonds in protein structures, and the “Complete collection” which in addition to that contains 4748 variants describing the rarely occurring disulfides. The details of constructing the 3D-motif libraries are provided in the Yosshi publication.
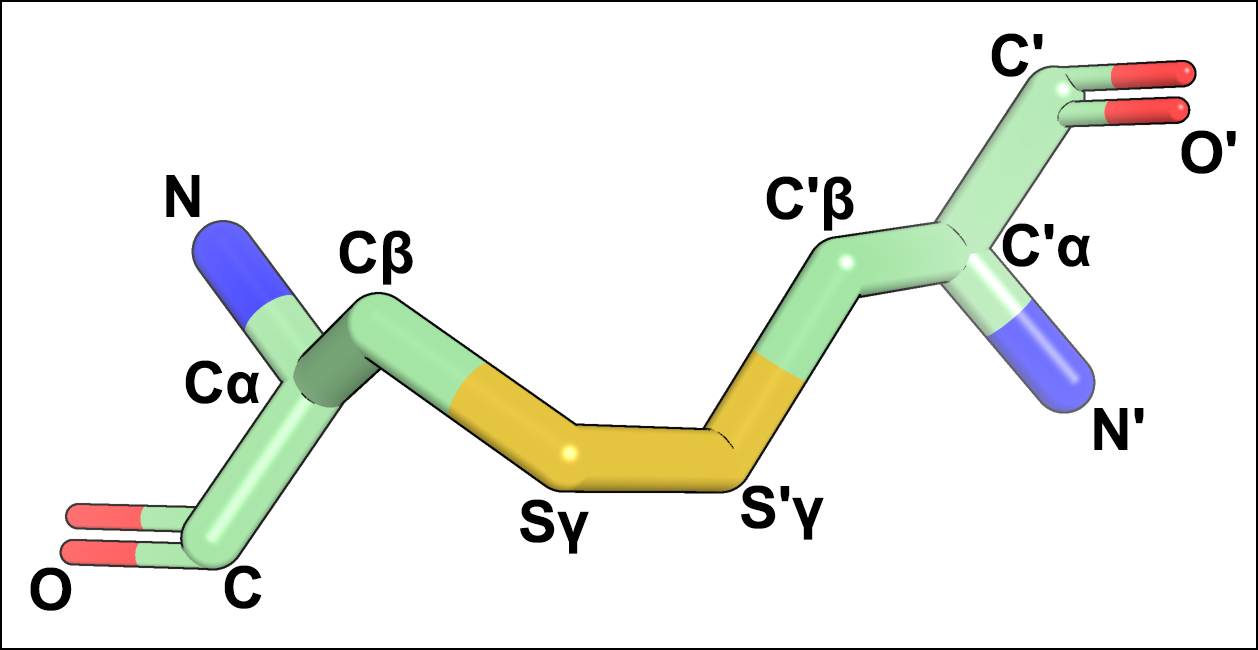
At the 3D-motif analysis step each pair of candidate positions in the query protein structure which passed the “First tier filter” and was further selected by the bioinformatic analysis (i.e., both positions are occupied by Cysteines in at least one homolog) would be confirmed as a promising site for S-S bond formation if it matches with at least one 3D-motif from the selected collection.
Two statistical models for the 3D-motif analysis and selection of the most promising sites in the query protein structure have been proposed: "Flexible" and "Rigid".
| Selected library of 3D-motifs | Default statistical model | μ, Å | σ, Å | Threshold X Å, where P(x>X)=0.05 |
| Core collection | Flexible | 0.39 | 0.19 | 0.70 |
| Complete collection | Rigid | 0.16 | 0.07 | 0.28 |
The Flexible statistical model is based on the bioinformatic analysis of homologous protein structures with different disulfide connectivity to take into account the flexibility of a pair of non-bonded amino acid residues which can form an S-S bridge upon mutation to Cysteines. This model was trained by comparing the disulfide bonds C1-C2 with their non-bonded equivalences E1-E2 in homologous proteins (see below). Under this “Flexible” model, a pair of positions in the query protein structure would be confirmed as a promising site for S-S bond formation if it matches with at least one 3D-motif from the select library so that both RMSD values corresponding to two pairs of superimposed backbone atoms are within X=0.70 Å, which corresponds to a p-value of P(x>X)=0.05 of a normal distribution with μ=0.39 Å and σ=0.19 Å, or rejected otherwise.
The Rigid statistical model has been proposed based on the analysis of 273 clusters incorporating typical S-S bond configurations produced by the DBSCAN machine-learning clustering algorithm from the analysis of 16956 disulfide variations (the details are provided in the Yosshi publication). Under this “Rigid” model, a pair of positions in the query protein structure would be confirmed as a promising site for S-S bond formation if it matches with at least one 3D-motif so that both RMSD values between two pairs of superimposed backbone atoms are within X=0.28 Å, which corresponds to a p-value of P(x>X)=0.05 of a normal distribution with μ=0.16 Å and σ=0.07 Å, or rejected otherwise.
In the "Flexible" mode the "Flexible" statistical model is loaded to run the 3D-motif analysis using the "Core collection" library. In the "Rigid" mode the "Rigid" statistical model is used together with the "Complete collection" library. In the "Custom" mode the user can choose one of the two libraries and set the statistical model manually. The details explaining the threshold selection and evaluation of structural filtration are provided in the Yosshi publication.
The relationship between the mean μ, standard deviation σ, p-value, and quantile (i.e., the Threshold - the exact RMSD cut-off between the 3D-motif and the candidate pair of two position in the query protein) can be probed by using the R-language. The R Project provides free software for Statistical Computing and is available for all operating systems (https://www.r-project.org/). To learn the quantile for a given statistics (i.e., mean, sd, and p-value of the normal distribution) use the qnorm command:
> qnorm(mean=0.39, sd=0.19, p=0.05, lower.tail = FALSE)
[1] 0.7025222
> qnorm(mean=0.39, sd=0.19, p=0.01, lower.tail = FALSE)
[1] 0.8320061
> qnorm(mean=0.39, sd=0.19, p=0.3, lower.tail = FALSE)
[1] 0.4896361
I.e., when the μ=0.39 Å, σ=0.19 Å, and p=0.05 the exact RMSD cut-off used by Yosshi is 0.7025222 Å
To learn the p-value that should be used in order to set the exact RMSD cut-off to a certain value, for a given statistics (i.e., mean, standard deviation of the normal distribution) use the pnorm command:
> pnorm(mean=0.39, sd=0.19, q=0.7025222, lower.tail = FALSE)
[1] 0.05
> pnorm(mean=0.39, sd=0.19, q=0.5, lower.tail = FALSE)
[1] 0.2813123
I.e., to set the exact RMSD cut-off used by Yosshi to 0.5 Å you can use μ=0.39 Å, σ=0.19 Å, and p=0.2813123.
Thresholds for structural filtration
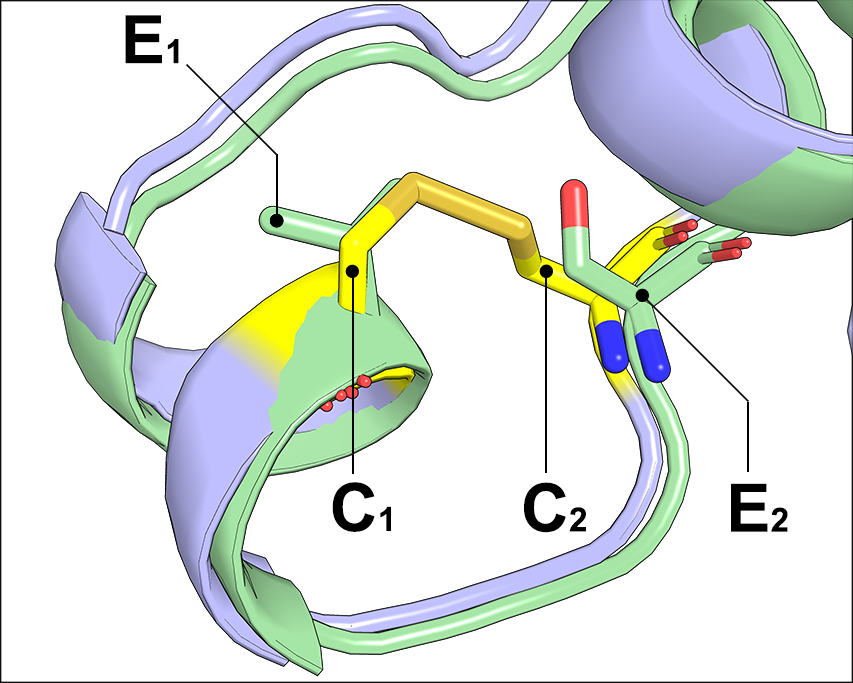
The Yosshi workflow consists of bioinformatic analysis to search for pairs of Cysteine residues in sequences and structure of homologs, and structural filtration to evaluate whether introduction of the selected Cysteines at corresponding positions of the query structure can result in a formation of a disulfide bond. The previously reported disulfide engineering experiments have demonstrated that introduction of a crosslink into the query structure is likely to result in a considerable shift of the backbone atoms, and thus the candidate hot-spot positions cannot be expected to match the strict geometric constraints of an S-S bond. Therefore, to perform such a structural filtration it is important to take into account flexibility of a pair of non-bonded amino acid residues which can form an S-S bridge upon mutation to Cysteines. In this study a large non-redundant set of disulfide bonds (C1 and C2) and their non-bonded equivalences in structures of homologous proteins (E1 and E2) was collected by bioinformatic analysis of protein superfamilies according to the CATH classification. Geometry constraints of backbone atoms in the collected non-bonded residue pairs were further studied and used to set the structural filtration thresholds within the Yosshi workflow, in particular, to train the "Flexible" statistical model for the 3D-motif analysis (see above). The details of this analysis are provided in the Yosshi publication.
Provide HTML links to PDB, UniProt, and BacDive

The first output of Yosshi is a list of pairs of positions in the structure of the query protein that can form a disulfide bond assuming both residues are mutated to cysteines, or that already are occupied by cysteines which can form a crosslink. The second output is a list of homologs of the query protein, which contain cysteines in equivalent positions for each pair of such residues. As an option, the user can request the web-server to provide the HTML links to PDB, Uniprot, and BacDive (Bacterial Diversity Metadatabase) databases for each selected homolog to facilitate further expert analysis. Such HTML links will be included into the downloadable text version of the Yosshi annotation and appear as hyperlinks at the on-line analysis page:

Click here to enlagre
This feature does not influence the main output of the web-server but facilitates further analysis of homologs by providing easy-to-use one-click access to a detailed information about the respective proteins in PDB or UniProt, and about their source organisms in the BacDive.
For this feature to work, the name string of each protein in the multiple sequence alignment FASTA file should contain the respective information (i.e., PDB or UniProt access code and title of source organism) in the appropriate format, as described below. The Mustguseal web-server will attempt to automatically prepare protein names in the correct format when constructing the alignment. The PDB/UniProt accession numbers are always included into the Mustguseal alignment; however, recognition of the titles of source organisms can be ambiguous or these titles may be incomplete (e.g., Streptomyces sp.) and will be provided "as is". Alternatively, if the multiple alignment is created by the user (i.e., without the Mustguseal), the correct format of protein names is responsibility of the user. If protein names fail to comply with the required format the HTML links would not be created. In such a case, the output created with this feature enabled and disabled would be identical.
Due to these input format requirements the feature is disabled by default, and would be automatically activated if the input multiple alignment is submitted to Yosshi from Mustguseal.
Please understand that at present this feature has some minor limitations. In particular, a link to either PDB or UniProt (i.e., not both) will be provided for each protein, and a link to the BacDive's search engine will be provided for all proteins whose name string in the multiple alignment file contained the name of a source organism, i.e., even a human protein would have such a link to the Bacterial Diversity Metadatabase and would, obviously, return a "Sorry, nothing found" statement. This feature will be improved in future releases of Yosshi to provide the links to both PDB and UniProt, and provide the BacDive links only for those proteins whose source organisms are actually described in that database. Despite some limitations the feature works and sends a clear message to the user, in particular, that the BacDive can help to quickly provide information on a particular bacterial source (e.g., see the Demo example for subtilisin E from Bacillus subtilis here).
Format requirements for compatibility with the "Provide HTML links to PDB, UniProt, and BacDive"feature:
The PDB codes should be provided to Yosshi in the following format within the protein name in the multiple alignment FASTA file: [ID]_[PDBC]_[chain], where [ID] is any number, [PDBC] is the 4-digit PDB code, and [chain] is a 1-character chain identifier, e.g.:
>122_3tec_E
YTP-N-DPY-FSSRQYGPQKIQAP
QAWDIAEGSG--AKIAIVDTGVQS
NHP---DLAGKVV-----------
For this example, the following HTML link to the PDB will be automatically created:
https://www.rcsb.org/structure/3tec
The UniProt AC codes should be provided to Yosshi in the following format within the protein name in the multiple alignment FASTA file: [AC]=[anyinfo], where [AC] is the UniProt AC code containing 6-to-10 characters, e.g.:
>P08594=AQL1_THEAQ_Aqualysin1_OS=Thermus_aquaticus_OX=271_GN=pstI_PE=1_SV=2
QSPA---PWGLDRIDQRDLPLSNS-Y
TYTATGRGVNVYVID--TGIRTTHRE
FGGRARV-----GYDALGG-------
For this example, the following HTML link to the UniProt will be automatically created:
https://www.uniprot.org/uniprot/P08594
The source organism title should be provided to Yosshi in the following format within the protein name in the multiple alignment FASTA file: [anyinfo]OS=[genus]_[species][anyinfo], where [genus] and [species] is the two-name of the Binomial Naming System, e.g.:
>P08594=AQL1_THEAQ_Aqualysin1_OS=Thermus_aquaticus_OX=271_GN=pstI_PE=1_SV=2
QSPA---PWGLDRIDQRDLPLSNS-Y
TYTATGRGVNVYVID--TGIRTTHRE
FGGRARV-----GYDALGG-------
For this example, the following HTML link to query the BacDive's search engine will be automatically created:
https://bacdive.dsmz.de/search?search=Thermus+aquaticus