Preparing the input data for Yosshi analysis
To use Yosshi you have to submit (1) a multiple alignment of proteins in the FASTA format and (2) a representative protein (i.e., the query) structure in the PDB format which should correspond to one of the proteins in the multiple alignment. This input can be prepared automatically and fully on-line by the Mustguseal web-server.
- Overview of the input data
- Automatic preparation of the input data by Mustguseal
- Using protein structures not (yet) in the PDB as queries to the Yosshi and Mustguseal
- The feature "Provide HTML links to PDB, UniProt, and BacDive"
- Guidelines for manual preparation of the input data
- Dealing with the multiple-chain proteins
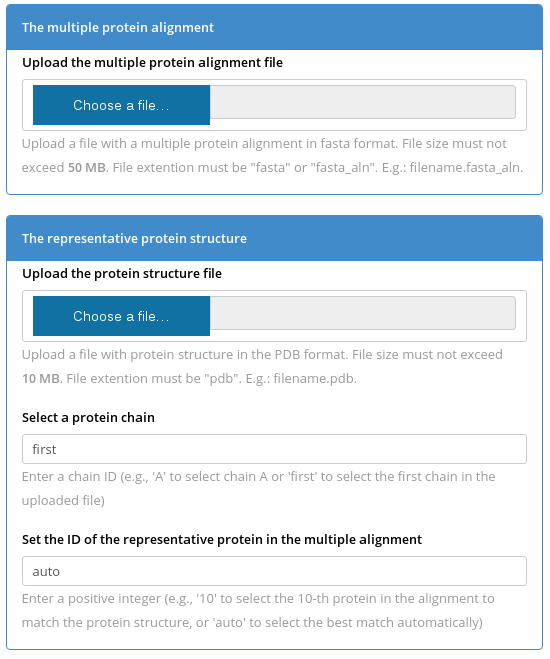
You have to submit two files to the Yosshi server - a multiple protein alignment in FASTA format and a representative protein (i.e., the query) structure in PDB format. To select a protein chain for the bioinformatic analysis use the "Select a protein chain" field - you can type in a particular chainID (case sensitive) or leave it to "first" to select the first chain that appears in the PDB file. The representative protein structure should match one protein in the sequence alignment. You can set the "Set the ID of the representative protein in the multiple alignment" field to "auto" for the server to automatically match the PDB structure with the alignment to select the best pairwise sequence superimposition, or set a number corresponding to the order of appearance of the representative protein in the multiple sequence alignment file (the numbering starts from "1").
Automatic preparation of the input data by Mustguseal
The input to Yosshi can be automatically prepared by the sister web-server Mustguseal via a web-interface. Mustguseal can automatically construct large structure-based sequence alignments of functionally diverse protein families that include thousands of proteins based on all available information about their structures and sequences in public databases. The alignment automatically constructed using that public web-server can then be automatically submitted to the Yosshi web-server in one click.
Step 1: Choose the query protein. Choose the query protein based on your particular task and primary interest. It can be the target protein selected for the further experimental design, the most studied member of the superfamily, or a protein which you are the most familiar with.
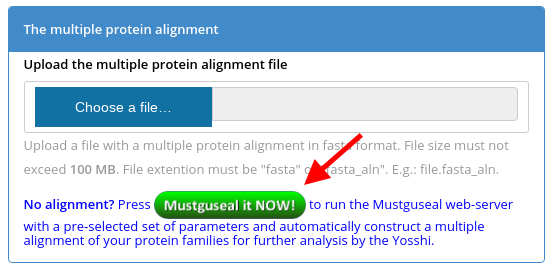
Step 2: Automatically construct a large alignment of diverse protein families. At the Yosshi submission page press the "Mustguseal it NOW" button (see a screenshot below) or simply press this one -  . This will redirect you to the Mustguseal web-server and load a pre-selected set of parameters (i.e., the Scenario 3).
. This will redirect you to the Mustguseal web-server and load a pre-selected set of parameters (i.e., the Scenario 3).
Enter the PDB ID and chain ID of your query protein (e.g., PDB 1SCJ chain A of subtilisin E from Bacillus subtilis, as discussed here) in the "Query protein" box and press "Submit" to automatically construct a large structure-guided sequence alignment of proteins with high structural but low sequence similarity to your query protein.

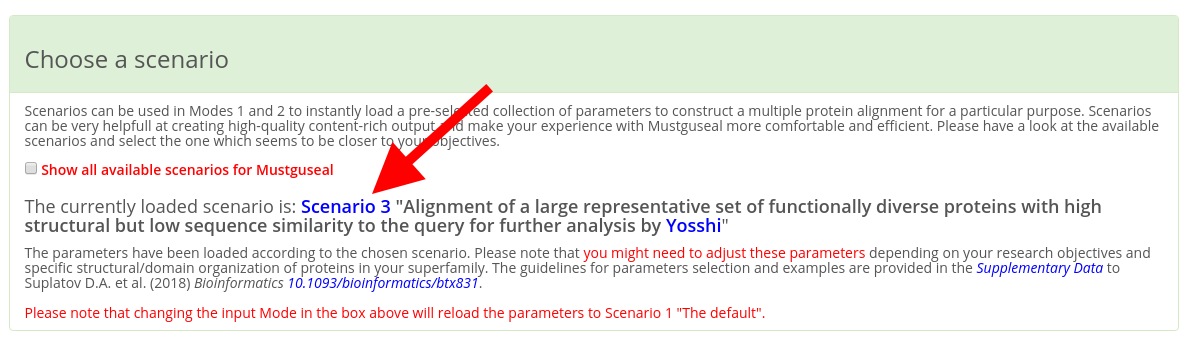
IMPORTANT! When making a submission to Mustguseal check that the "Choose a scenario" box states that the parameters have been set up according to the "Scenario 3" (see a screenshot below). If you have entered the Mustguseal submission page not from the Yosshi web-page than other scenarios might be loaded. It that case set the "Scenario 3" manually by checking the corresponding radio button. The parameters in the "Scenario 3" were chosen to optimize the performance of Mustguseal at constructing large and diverse alignments of protein families for further analysis by Yosshi. Please note that you might need to adjust these parameters depending on your research objectives and specific structural/domain organization of proteins in your superfamily. The guidelines for parameters selection and examples are provided in the Supplementary Data to the Mustguseal publication.
Step 3: Automatically transfer your data from Mustguseal to Yosshi. A "Results" button will appear at the Mustguseal progress log page upon the completion of your task. Press that button to access the Mustguseal "Results" page. At that page page scroll down and press the "Submit to Yosshi" button. This will automatically transfer the collected sequence and structural data to Yosshi.

Step 4: Start Yosshi. Press "Submit" button to start the Yosshi routine. When your task is processed, press "Results" to access the results and the download section, and press "Analysis" to load the interactive on-line analysis tools.
Using protein structures not (yet) in the PDB as queries to the Yosshi and Mustguseal
Can a protein structure not yet in the PDB (or a homology model) be used as a query to the Yosshi? Yes, it can. Upload the corresponding file in the PDB format to Yosshi using the standard File Upload dialog box, and then upload the corresponding multiple alignment that contains this protein. If you do not have such an alignment - see below.
Can a protein structure not yet in the PDB (or a homology model) be used as a query to the Mustguseal to automatically construct the alignment for further processing by the Yosshi? No, it can not. If your protein structure is new and not (yet) in the PDB (or a homology model) you have two options: (1) use a PDB of a close homolog to construct the alignment by Mustguseal and then upload the new protein structure in the PDB format together with the alignment to Yosshi (i.e., the PDBeFOLD web-server can be used to search for structurally similar proteins in the PDB to select such a homolog with a known 3D-structure) or (2) create and submit a custom-built core structural alignment to Mustguseal to create a corresponding structure-guided sequence alignment in Mode 2 (as explained here), and then submit it to Yosshi together with the new protein structure in the PDB format.
The feature "Provide HTML links to PDB, UniProt, and BacDive"
The user can request the web-server to provide the HTML links to PDB, Uniprot, and BacDive (Bacterial Diversity Metadatabase) databases for each homolog selected by the bioinformatic analysis to facilitate further expert analysis in the output files and at the on-line analysis page. For this feature to work, the name string of each protein in the multiple sequence alignment FASTA file should contain the respective information (i.e., PDB or UniProt access code and title of source organism) in the appropriate format. The Mustguseal web-server will attempt to automatically prepare protein names in the correct format when constructing the alignment. For multiple alignments created by the user (i.e., without the Mustguseal), the correct format of protein names for compatibility with the "Provide HTML links to PDB, UniProt, and BacDive" is responsibility of the user. This feature and the required input format are discussed at the Yosshi parameters page.
Guidelines for manual preparation of the input data
Alternatively, you can prepare the input manually. The multiple sequence alignment should represent the desired diversity among the protein families of interest (i.e., contain homologous proteins with different function, stability and regulation). The representative protein structure is expected to correspond to a protein in the sequence alignment. Choose the representative protein based on your particular task and primary interest. It can be the target protein selected for the further experimental design, the most studied member of the superfamily, or a protein which you are the most familiar with. You should always aim at submitting a representative PDB structure that corresponds to a protein in the alignment with at least 95% pairwise sequence identity. If structural information is not available for all proteins in your alignment then you could use the structure of a very close homolog from the PDB database or build a 3D model of the representative protein based on the available structural data using the homology modeling (e.g., with the help of the highly capable Modeller software). For multiple alignments created by the user (i.e., without the Mustguseal), the correct format of protein names for compatibility with the "Provide HTML links to PDB, UniProt, and BacDive" is responsibility of the user. This feature and the required input format are discussed at the Yosshi parameters page.
General requirements for the input files
The input multiple alignment:
- should contain protein amino acid sequences;
- should contain at least six proteins;
- should be an alignment (i.e., not just sequences, but aligned sequences, i.e., "sequences with gaps");
- the "-" character should be used for a gap;
- the special characters in the protein names are not allowed and will be automatically substituted for "_";
- the very long protein names will automatically truncated to the first 100 characters;
- the special characters in the protein sequences are not allowed and will be automatically substituted for gaps;
- should be in the FASTA format (not ClustalW, not Phylip, etc.). If you do not know what is the format of your alignment - submit it to Yosshi and you`ll find out. If you have your alignment in the wrong format use a sequence format converter, e.g., sequenceconversion.bugaco.com;
The input protein structure:
- should contain the coordinates of amino acids atoms of one protein;
- should be in the PDB format;
- should correspond to (i.e., ideally should be 100% identical to) one protein sequence in the multiple alignment;
- may not be 100% identical to any protein sequence in the multiple alignment. The preprocessing script will automatically select the representative sequence from the multiple alignment by the best pairwise match between your PDB structure and any sequence in the alignment. All inconsistencies between the representative protein structure and sequence will be removed. You should always aim at submitting a representative PDB structure that corresponds to a protein in the alignment with at least 95% pairwise sequence identity. You will be allowed to proceed with up to 50% sequence similarity between the two, however, this may cause errors during the bioinformatic analysis.
- should contain all chains of the biological unit (e.g., A, B, C) even if the multiple alignment contains the sequences of only one chain (e.g., A);
- may contain heteroatoms. All non-protein atoms (e.g., of a substrate) should have the HETATM prefix in the PDB file. Non-canonical amino acids will be automatically changed to the canonical equivalents (i.e., SME/MSE to MET). Ligands, cofactors, solvent and other instances will not be used for the bioinformatic analysis but will be used to prepare the graphical output and can help with the interpretation of functional and regulatory significance of the predicted co-evolving positions.
Dealing with the multiple-chain proteins
Your protein contains multiple chains and you are interested in the analysis of disulfides which crosslink different chains (i.e., inter-chain/inter-subunit S-S bonds)? This topic is discussed on a separate page - press here for a redirect.