Open-access Mustguseal platform for bioinformatic analysis in computational enzymology
Comparative analysis of homologous proteins in a functionally diverse superfamily is a valuable tool at studying structure-function relationship, but represents a methodological challenge. We have developed an open-access platform uniting original on-line methods to study the structure-function relationship in proteins, to select the most promising hot-spots for implementation of novel functions, improvement of stability and evolvability of useful proteins/enzymes, and to design of their selective modulators.
THIS WEB-SITE/WEB-SERVER/SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS OR HARDWARE OWNERS OR WEB-SITE/WEB-SERVER/SOFTWARE MAINTEINERS/ADMINISTRATORS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE WEB-SITE/WEB-SERVER/SOFTWARE OR THE USE OR OTHER DEALINGS IN THE WEB-SITE/WEB-SERVER/SOFTWARE. PLEASE NOTE THAT OUR WEBSITE COLLECTS STANDARD APACHE2 LOGS, INCLUDING DATES, TIMES, IP ADDRESSES, AND SPECIFIC WEB ADDRESSES ACCESSED. THIS DATA IS STORED FOR APPROXIMATELY ONE YEAR FOR SECURITY AND ANALYTICAL PURPOSES. YOUR PRIVACY IS IMPORTANT TO US, AND WE USE THIS INFORMATION SOLELY FOR WEBSITE IMPROVEMENT AND PROTECTION AGAINST POTENTIAL THREATS. BY USING OUR SITE, YOU CONSENT TO THIS DATA COLLECTION.
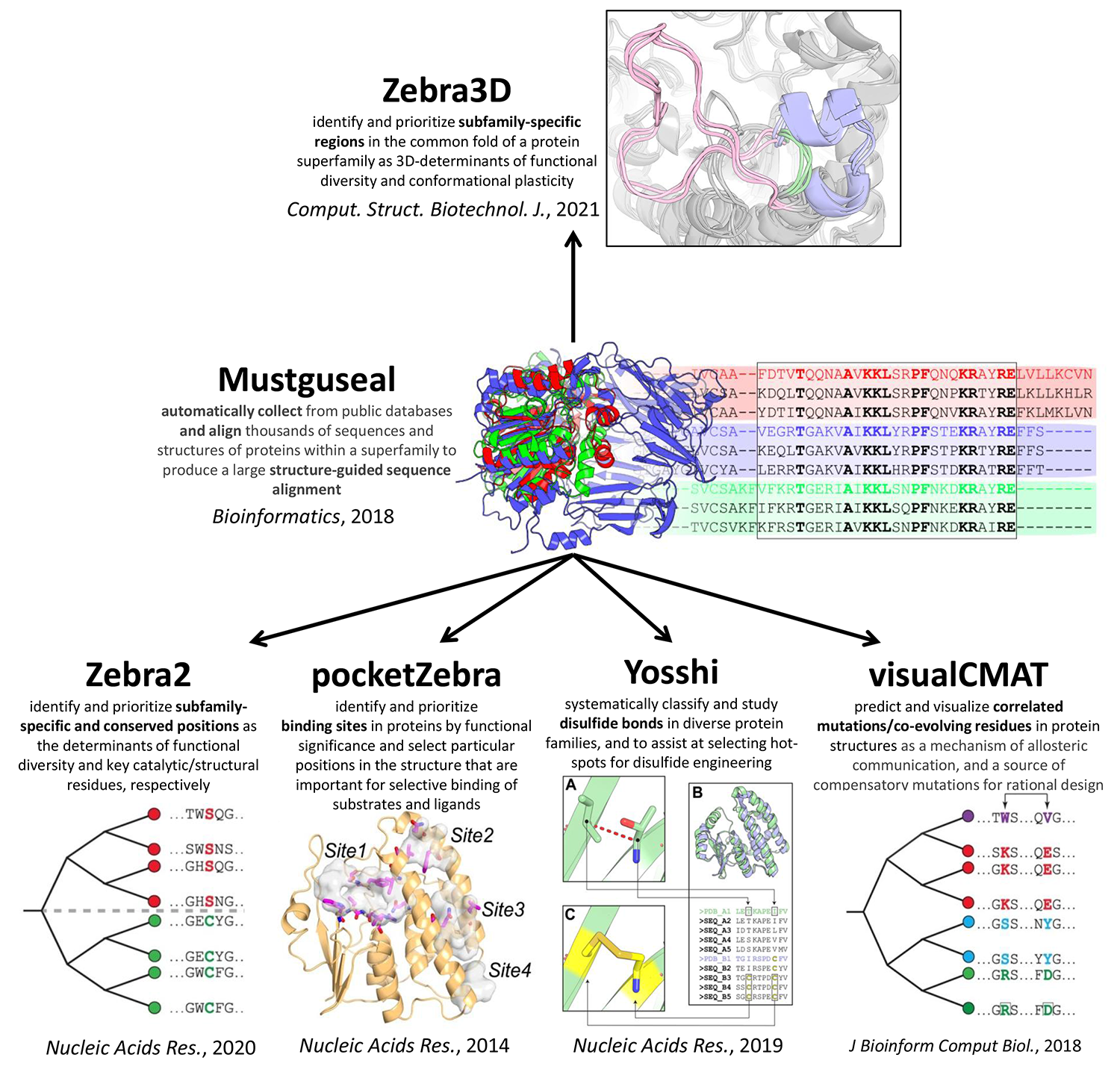
Mustguseal web-server can automatically collect and align thousands of homologous protein sequences and structures [Bioinformatics, 2018, DOI:10.1093/bioinformatics/btx831]
Five sister web-methods are available for subsequent bioinformatic analysis of the collected data:
- Zebra web-server to identify and prioritize conserved and specific positions in a functionally diverse superfamily and to select hot-spots for rational design of the query protein [Nucleic Acids Res., 2020, DOI:10.1093/nar/gkaa276];
- Zebra3D tool for bioinformatic analysis of 3D-determinants of functional diversity and function-related conformational plasticity in protein superfamilies using machine learning [Comput. Struct. Biotechnol. J., 2021, DOI:10.1016/j.csbj.2021.02.005];
- the pocketZebra web-server to identify and rank binding sites in proteins by their functional significance and to select particular positions in the structure important for selective binding of substrates/inhibitors/effectors [Nucleic Acids Res., 2014, DOI:10.1093/nar/gku448];
- the visualCMAT web-server to select and interpret correlated mutations/co-evolving residues in protein structures [J Bioinform Comput Biol., 2018, DOI:10.1142/S021972001840005X];
- Yosshi web-server to classify and study disulfide bonds in protein families as well as to select hot-spots for disulfide engineering [Nucleic Acids Res., 2019, DOI:10.1093/nar/gkz385].
Integration of these bioinformatic web-tools provides an out-of-thebox easy-to-use solution, first of its kind, to systematically analyze all the available sequence and structural data related to a protein superfamily, thus promoting the value of bioinformatics for protein engineering and drug discovery.
Practical guide: Suplatov D., Sharapova Y., Švedas V. (2021) Mustguseal and Sister Web-Methods: A Practical Guide to Bioinformatic Analysis of Protein Superfamilies. In: Katoh K. (eds) Multiple Sequence Alignment. Methods in Molecular Biology, vol 2231. Humana, New York, NY. DOI: 10.1007/978-1-0716-1036-7_12.
A brief overview of the Mustguseal platform:
| Download PDF file (2.3 MB) |
This work is being funded by the Russian Foundation for Basic Research [#18-29-13060] and carried out using the HPC computing resources at the Lomonosov Moscow State University supported by the project RFMEFI62117X0011.