Input to Mustguseal
Mustguseal can be used to build alignments of the selected protein families or collect and superimpose a large set of related proteins within a superfamily. The scope of the final alignment is defined by the diversity of representative proteins in the core structural alignment which can be created on-site or submitted by the user. There are three ways of submitting a task to the Mustguseal. Mode 1 is the default, fully automated, and the easiest way to obtain your alignment. Modes 2 and 3 provide an opportunity to refine the alignment by editing the components it is built from. The three modes provide a full control over the alignment construction process. Users are advised to use the Mode 1 as the default and then switch to Mode 2, and then to Mode 3, if necessary. The special-purpose Mode 4 is available to construct only the core 3D-structural alignment. This page describes how you can use Mustguseal to build a large alignment of your protein families for a particular purpose.
There are three input modes and the results claim form available on the Mustguseal submission page:
- Mode 1: Submit a query protein
- Mode 2: Submit a core structural alignment
- Mode 3: Submit a core structural alignment and results of sequence similarity search
- Mode 4: Submit a query protein to construct only the core 3D-structural alignment
- Claim your results by TaskID
Mode 1: Submit a query protein
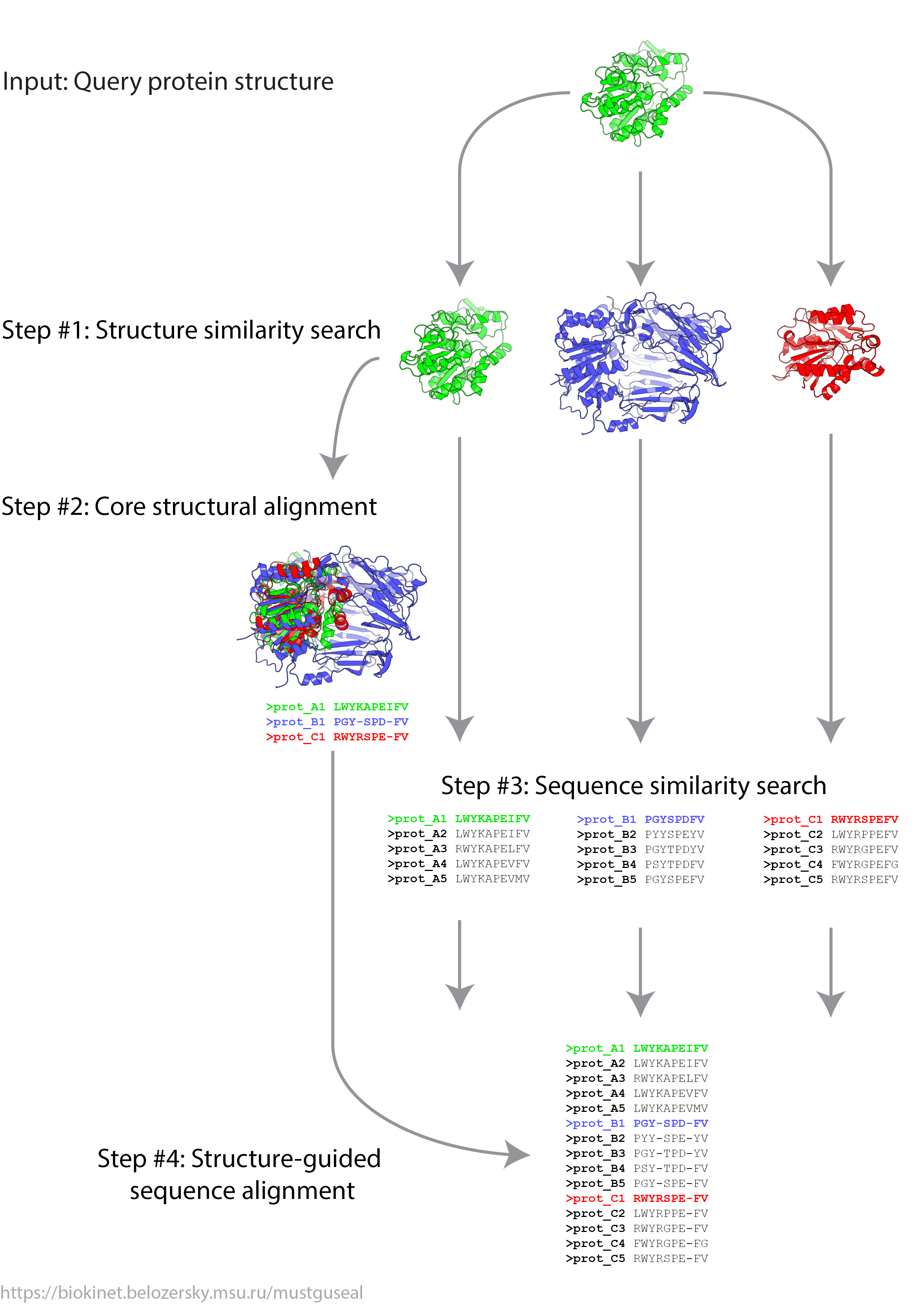
In Mode 1 steps 1 - 4 of the Mustguseal protocol are executed. This mode will automatically collect and align all structures and sequences of proteins homologous to your query and produce the final structure-guided sequence alignment. Users are advised to use this mode as the default.
Guideline for the query selection. In Mode 1 you submit PDB and chain IDs of a query protein. Select the query protein based on your particular task and primary interest. It can be the target protein selected for the further experimental design, the most studied member of the superfamily, or a protein which you are the most familiar with. If structure of the selected protein is made up of identical subunits (e.g., homotetramer) then choice of the chain is arbitrary. If structure of the selected protein is made up of different subunits (e.g., heterodimer) you should start from submitting the ID of a chain which is primary to protein function (e.g., contains the catalytic residues and the active site of an enzyme). Generally speaking, if structure of the selected protein is made up of different subunits you should build a separate alignment for each chain. Please note that the chain ID is case sensitive (i.e., 'A' and 'a' will be considered as different inputs).
Policy on incomplete PDB structures. Incomplete structures are a common case in the PDB database. The most prominent problems are modified/non-standard residues annotated as heteroatoms and missing flexible loops. When running in Mode 1 Mustguseal automatically applies the following modifications to the PDB files:
- All selenomethionine are converted to methionines and considered for the structural alignment;
- All heteroatoms, which contain a "CA" atom and are listed in the SEQRES field of the corresponding PDB file will be considered for the structural alignment and will appear as "X" in the final alignment and sequence representation of the structural alignment;
- All heteroatoms, which contain a "CA" atom but are not listed in the SEQRES field of the corresponding PDB file will have the "CA" atom renamed to "CX" and will neither be considered for the structural alignment nor will appear in the final alignment;
- All heteroatoms, which do not contain a "CA" atom will neither be considered for the structural alignment nor will appear in the final alignment.
All modifications are printed to the log file, e.g.:
Warning: Non-standard residue LLP A 140 contains atom CA and is listed in the SEQRES field of PDB file 80_3lul_A.pdb, and thus will appear as X in the core structural alignment
Info: Renaming selenomethionine(s) to methionine(s) in PDB file 80_3lul_A.pdb
Info: Processing PDB file 80_3lul_A.pdb ... done
Warning: Renaming atom CA to CX in heteroatom/ligand 4MV B 420 in PDB file 144_2cog_B.pdb
Info: Processing PDB file 144_2cog_B.pdb ... done
You should check if the PDB structures which were used to build the core structural alignment in Mode 1 are incomplete (you can download the respective data at the Results page), and if they are - manually evaluate the importance of the missing/modified regions for your study. You could build the complete models of your protein structures using the molecular modeling software (e.g., Modeller), align them locally on your computer or supercomputer, and then submit this user-defined core structural alignment to the server in Mode 2 or Mode 3.
Policy on non-standard amino acid sequence codes. The B, J, and X are allowed in the protein amino acid sequences. The Z is converted to a gap. The U (i.e., selenocysteine) is converted to C, and O (i.e., pyrrolysine) is converted to K.
Mode 2: Submit a core structural alignment
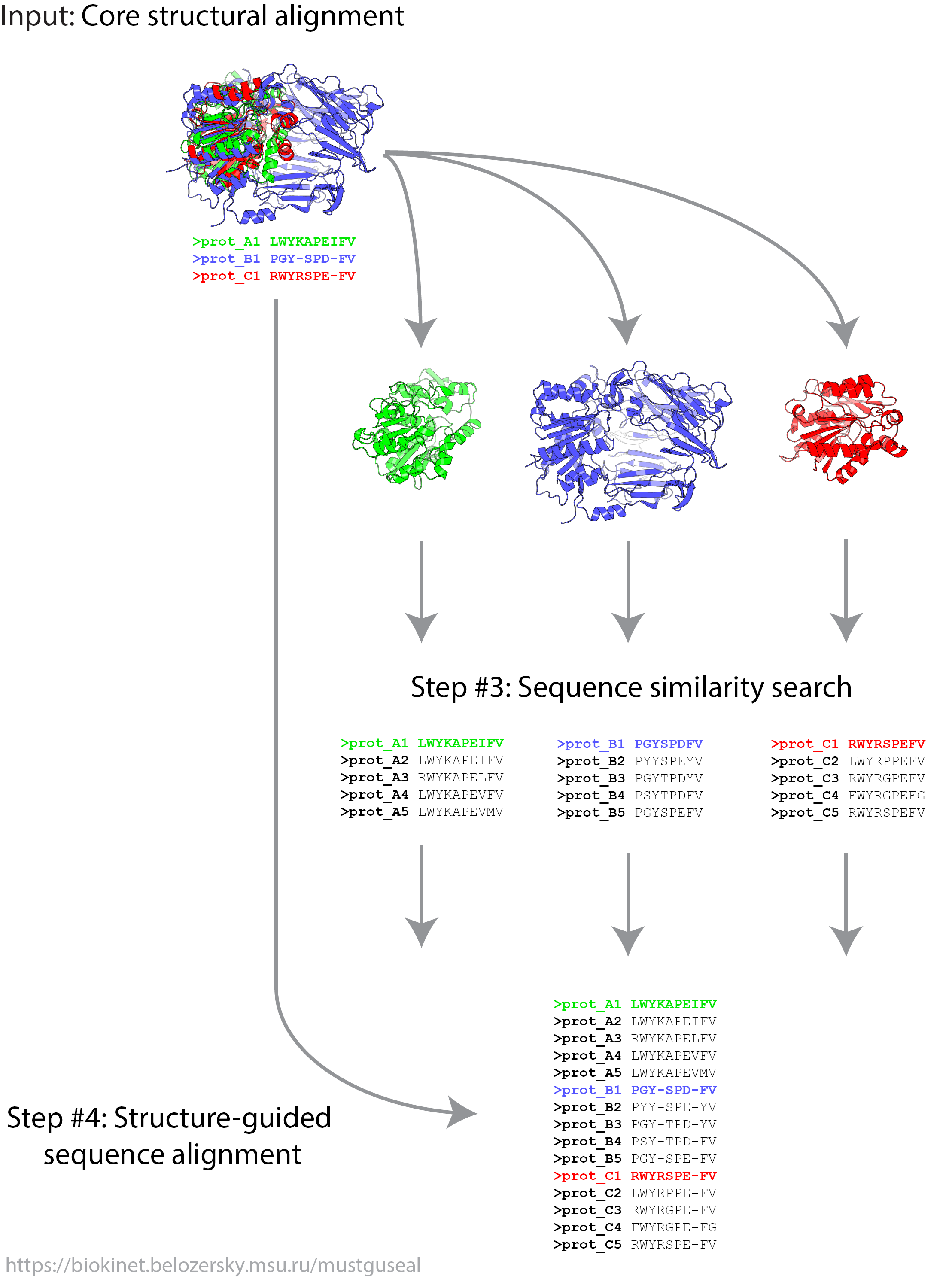
This mode provides an opportunity to submit a user-defined core structural alignment to construct a structure-guided sequence alignment of the selected protein families and their closest homologs. In Mode 2 steps 3 and 4 of the Mustguseal protocol are executed.
Structural alignment of a representative set of homologous proteins is the core of the Multiple Structure-Guided Sequence Alignment. It is important that proteins in this core structural alignment represent the desired diversity among the protein families of interest for a particular research objective. The user may wish to include different structures in the core structural alignment (i.e., different from what has been selected automatically by the server in Mode 1) or alter in any way the alignment itself (e.g., by manually editing local amino acids superposition).
File Format Requirements in Mode 2. The sequence representation of the core structural alignment (i.e., not the 3D coordinates but the fasta sequence file) should be submitted in Mode 2. One text file (flatfile) with the '.fasta_aln' extension has to be submitted with sequences in the FASTA format. Examples: corealn.fasta_aln, but not corealn.fasta or corealn.something.fasta_aln. If the file describes only one protein then its sequence should be without gaps. If the file describes two or more proteins then their sequences must be aligned - i.e., the total length of protein amino acid sequence plus gaps must be the same for all proteins in the file. Protein names should not exceed 100 characters in length and should not contain special characters. Protein sequences should not contain the 'Z' character as well as any special characters or numbers.
How to build a custom core structural alignment for Mode 2?
A user-defined 3D-alignment can be built from a selected set of protein structures on a local computer/supercomputer or a third party web-server and then submitted to the Mustguseal server in Mode 2. This provides an opportunity to construct a structure-guided sequence alignment of the selected protein families and their closest homologs. The general guideline for customizing the core structural alignment follows:
- Run the structure similarity search in Mode 1 to automatically collect a set of structures related to your query (i.e., the protein of primary interest). Submit a query protein in Mode 1, download the archive with the structure similarity search results, and check the
superimpose.listfile for the full list of structural similarities with the query. For each pairwise match the PDB annotation will be provided. The 95%-non-redundant set (by pairwise sequence similarity) of protein structures will be provided in theresults_nr95/folder (see the Explanation of the Output for more details). Select representative proteins for the core structural alignment based on your particular task and primary interest. E.g., if you want to compare homologous enzymes with amidase and lipase activities, then pick the structures from the non-redundant set with the respective annotation;
- Alternatively, to select representative proteins you can use the popular PDBeFOLD web-server (http://www.ebi.ac.uk/msd-srv/ssm/). This public on-line method implements the SSM algorithm and can be used to run the structure similarity search versus the complete PDB database. One benefit of such approach is that you can submit your own PDB file (e.g., a structure which is not yet available in the PDB database) to that web-server to collect proteins structurally similar to your query, and then create the core structural alignment for further processing by the Mustguseal, as explained below;
- Create the structural alignment of the selected proteins - on your local computer or at the third-party web-server:
- You can use the MATT algorithm to compare remote evolutionary relatives (Menke et al., 2008). MATT searches for compatible pairs of fragments and permits structural allowances such as twists and translations, and demonstrates good performance in aligning distant relationships and length variations (Kalaimathy et al., 2011). MATT can be downloaded and installed locally or executed on the original web-server (http://matt.cs.tufts.edu/). MATT will produce the sequence representation of the core structural alignment (i.e., the '.fasta' file) which can be submitted to this server.
Example:
(1) Go to the MATT web-server;
(2) Enter the input PDB and chain IDs separated by space or comma: "1ev4:A 1f2e:A 1jlv:A 1fw1:A 1ljr:A 1lbk:A 1gwc:A 1axd:A 1eem:A", submit the task;
(3) Upon successful completion of the task you should be automatically redirected to the results page where your can download the output "alignment.fasta" file to your local computer;
(4) Rename the "alignment.fasta" to "alignment.fasta_aln";
(5) Submit the file to the Mustguseal server in the Mode 2.
NB: Sometimes the MATT server fails to load a particular PDB code (e.g.: "Error reading pdb//pdb3wwi, skipping"). To overcome this error download the PDB file manually from the PDB database and then upload it to the MATT server;
- You can use the PROMALS3D algorithm to compare remote evolutionary relatives (Pei et al., 2008). PROMALS3D can be downloaded and installed locally or executed on the original web-server (http://prodata.swmed.edu/promals3d/). PROMALS3D will produce the sequence representation of the core structural alignment (i.e., the '.fasta' file) which can be submitted to this server.
Example:
(1) Go to the PROMALS3D web-server;
(2) Enter the input PDB and chain IDs in the corresponding fields: "3wwi K", "4cmd B", "4uug A", "4ce5 A", submit the task;
(3) Upon successful completion of the task (can take some time) you should be automatically redirected to the results page. Find the "FASTA format alignment" section, press "Show", press right-click button on your mouse, select "Save as" option, choose the folder to store the file and enter the file name "anyname.fasta_aln". The alignment will be downloaded to your local computer;
(4) Submit the file to the Mustguseal server in the Mode 2;
Kalaimathy,S., Sowdhamini,R., and Kanagarajadurai,K. (2011). Critical assessment of structure-based sequence alignment methods at distant relationships. Brief. Bioinformatics, 12(2), 163-175
Menke,M., Berger,B., and Cowen,L. (2008). Matt: local flexibility aids protein multiple structure alignment. PLoS Comput Biol, 4(1), e10 Pei J., Kim B.H., Grishin N.V. (2008).
PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic acids res., 36(7), 2295-2300.
- You can use the MATT algorithm to compare remote evolutionary relatives (Menke et al., 2008). MATT searches for compatible pairs of fragments and permits structural allowances such as twists and translations, and demonstrates good performance in aligning distant relationships and length variations (Kalaimathy et al., 2011). MATT can be downloaded and installed locally or executed on the original web-server (http://matt.cs.tufts.edu/). MATT will produce the sequence representation of the core structural alignment (i.e., the '.fasta' file) which can be submitted to this server.
- You can use parMATT (https://biokinet.belozersky.msu.ru/parmatt) to build a customized core structural alignment from a large collection of user-selected protein structures on your local computing cluster or a supercomputer. The parMATT is a parallel MPI re-implementation of the MATT algorithm and is intended for distributed-memory systems (i.e., computing clusters and supercomputers hosting memory-independent computing nodes). parMATT can significantly accelerate the time-consuming process of building a large structural alignment. parMATT takes protein structures in the PDB format as input and produces structural alignment in both the PDB and FASTA formats, the latter being fully compatible with the Mustguseal Mode 2 input requirements.
Mode 3: Submit a core structural alignment and results of sequence similarity search
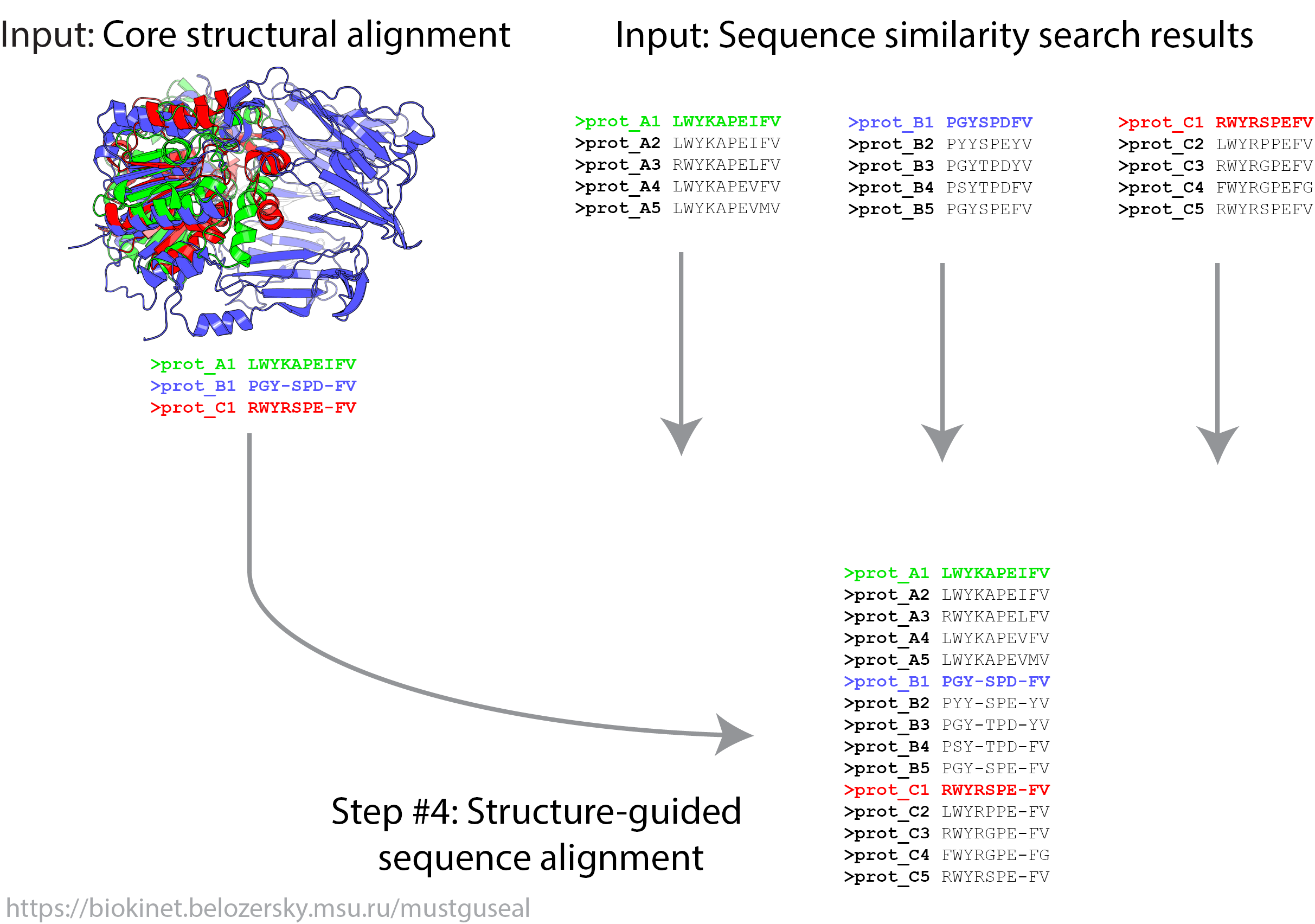
In Mode 3 steps 3 - 4 of the Mustguseal protocol are executed. This mode provides an opportunity to submit a user-defined core structural alignment as well as sequence alignment blocks of close homologs corresponding to each representative protein in the core structural alignment. User may alter in any way the results of sequence similarity search obtained automatically in Mode1 or Mode 2 (i.e. choose different proteins or change the way sequences are being superimposed within each group) and then submit all building blocks of the alignment in Mode 3. Please note that the sequence representation of the core structural alignment (i.e., not the 3D coordinates but the fasta sequence file) should be submitted in Mode 3.
File Format Requirements in Mode 3. Two file have to be submitted. The first file is the core structural alignment - see the File Format Requirements in Mode 2 above. The second file is an archive with sequence alignment files. This file must have the '.tgz' extension and correspond to a TAR+GZIP archive. To create this file use the command
tar czf upload.tgz folder_with_sequence_alignment_files
in Linux. In Windows use a free tool 7-zip to pack sequence alignment files into the '.tar.gz' archive and then manually change the file extension to '.tgz'. The archive must contain a collection of text files (flatfiles) with the '.fasta_aln' or '.final.fasta_aln' extension and sequences in the FASTA format. Examples: seqaln.fasta_aln or seqaln.final.fasta_aln, but not seqaln.fasta or seqaln.final.fasta or seqaln.something.fasta_aln. The structure of subcatalogs within the archive (i.e., the number of folders and their names) is not restricted given that each text file has a unique name. The technical requirements for the content of these files are the same as for the core structural alignment (see above). The names of sequence alignment files (e.g., pdb1.fasta_aln) must correspond to names of the representative proteins (e.g. pdb1) in the core structural alignment. Each sequence alignment file must include the respective representative protein and its amino acid sequence must be identical to that in the core structural alignment file.
Ensure that the archive contains exactly one sequence alignment file for each representative protein in the core structural alignment. Duplicate protein names within one alignment file are not allowed. Duplicate file names in different subfolders of the archive are not allowed. If a sequence alignment file is not available for a representative protein in the core structural alignment this would not result in an immediate termination and the Mustguseal will go on trying merge the available files. You should check for warning in the log file similar to an example below (use Ctrl-F to open the search field in your browser):
* ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * In Mode 3 you should submit a user-defined core structural alignment of representative proteins as well as an archive file with sequence alignment blocks of close homologs corresponding to each representative protein. The sequence alignment file corresponding to the representative protein 0_1p38_A must have a name 0_1p38_A.final.fasta, and this file seems to be missing. Consequently, in this task the sequences of proteins homologous to the representative protein 0_1p38_A will not be appended to the final alignment. Check the file names and submit a new task. Please note, that in some cases the filenames, protein names and sequences could be changed automatically during the Input Validation step if they contain special characters. Check the log for corresponding warning messages. * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION * ATTENTION *
This would mean that your archive did not contain the 0_1p38_A.final.fasta file and consequently the sequences of proteins homologous to 0_1p38_A were not appended to the final alignment. Check the file names and try again. Please note, that in some cases the filenames, protein names and sequences could be changed automatically during the Input Validation step if they contain special characters (this is a security feature to disarm unauthorized calls of service commands which could possess a threat to the server). In this case a warning would appear on the log:
Warning: The alignment file has been automatically modified ...
If you have a problem preparing the input in Mode 3 you should choose a query protein and submit it in Mode 1 (or submit the core structural alignment in Mode 2), then download the core structural alignment and the corresponding sequence alignments, and try using the automatically created alignments as a template to create your new input.
How to prepare the input for Mode 3?
The Mode 3 serves two purposes:
- It provides an opportunity to edit both the core structural alignment and the sequence alignment blocks (i.e. add/delete proteins or change the way sequences are being superimposed);
- It provides an opportunity to build a very large alignment based on the core structural superimposition of a large collection of remote evolutionary relatives (i.e., which represent a large protein superfamily);
In both cases the input for the Mode 3 should be prepared by implementing Modes 1 and 2 of the Mustguseal server. The core structural alignment constructed in the Mode 1 and the sequence alignment blocks constructed in Modes 1 and 2 can be downloaded to a local computer, edited/altered if necessary, combined with the output files from other submissions if necessary, and then submitted back to the server in the Mode 3.
E.g., a structural alignment of pdb1, pdb2, ... , pdb16 can be submitted in the Mode 2 to automatically prepare the corresponding 16 sequence alignment blocks, then a structural alignment of pdb17, pdb18, ... , pdb32 can be submitted as a different task in the Mode 2 to collect another 16 sequence alignment blocks, and then the complete structural alignment of pdb1, pdb2, ... , pdb32 and the 32 previously prepared sequence alignment blocks can be submitted in the Mode 3 to merge them together.
See the complete description of the Mode 3 and file format requirements, the Mustguseal Output for a detailed description of the Mustguseal output files, and the Mustguseal Performance for the description of current limitations on the input in the Modes 1, 2 and 3.
Mode 4: Submit a query protein to construct only the core 3D-structural alignment
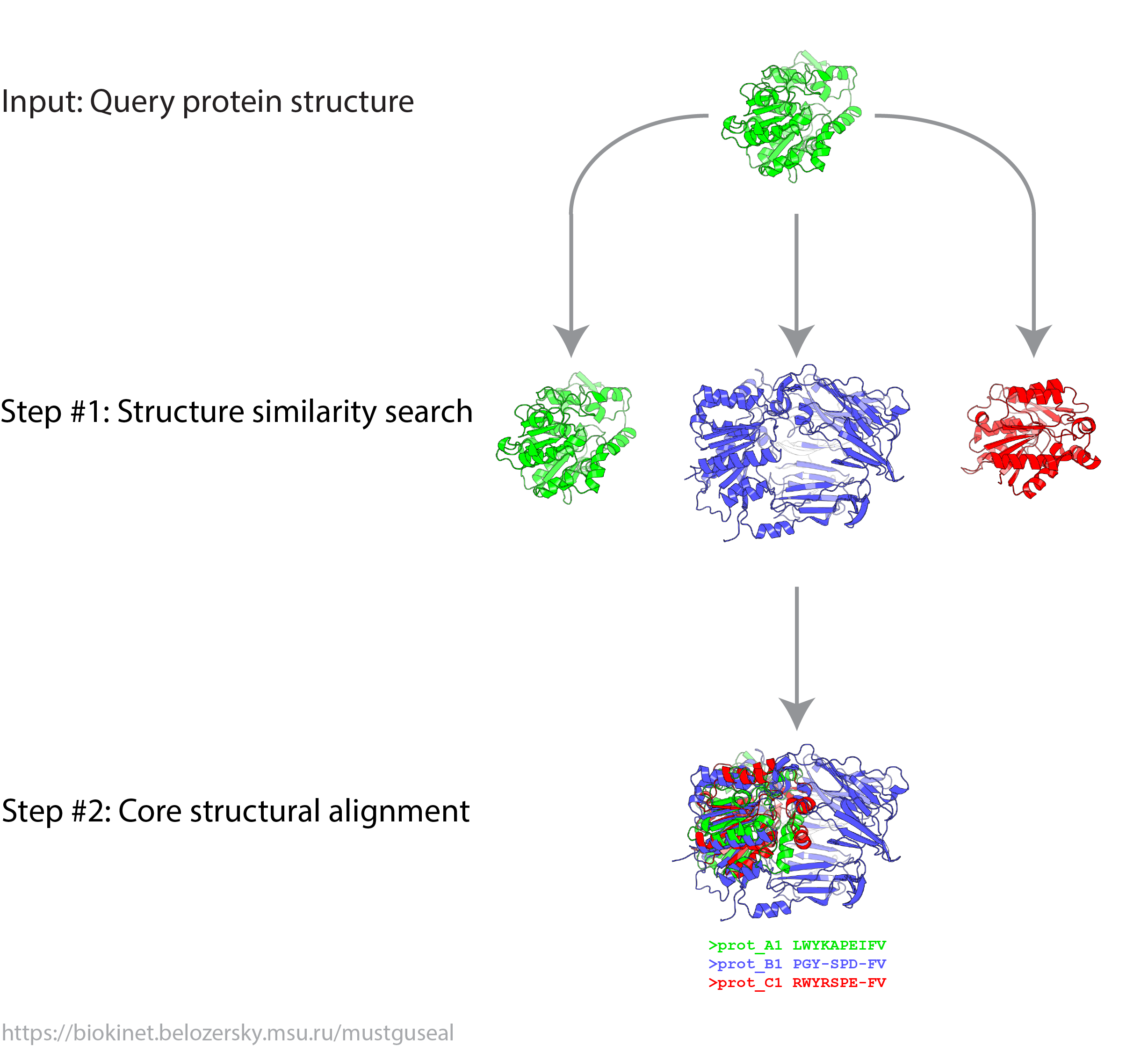
The special-purpose Mode 4 is available to collect PDB entries similar to the query and construct the corresponding 3D-structural alignment. The input to this mode is qualitatively similar to that in the Mode 1; however, only the steps 1 and 2 of the Mustguseal protocol will be carried out (i.e., the structure similarity search and subsequent 3D-alignment). The Mode 4 was specifically designed to assist the preparation of input data to the Zebra3D utility - a bioinformatic tool to systematically study 3D-structural diversity and specificity in protein superfamilies.
Claim your results by TaskID
The results and progress log of a previously submitted task can be accessed on entering a 16-symbol TaskID in the corresponding form available at the Mustguseal submission page.