
Informational and File Sharing Server
Modeling of Macromolecular Systems
Software
vsFilt: a tool to improve virtual screening by structural filtration of docking poses
Abstract of vsFilt: The ability of ligands to form specific interactions with the protein target, a characteristic of substrates and inhibitors, is a powerful structural criterion to identify potent binders among docked poses. Structural filtration of the raw output of virtual screening can help to recover the most promising ligands in the productive binding orientation for further analysis, but requires specialized software to automate the evaluation of large libraries. The vsFilt is the first open application for post-docking structural filtration, available as a web-server. The new tool is easy to use and configure to detect a wide range of interaction types that are known to be involved in molecular recognition, including hydrogen and halogen bonds, ionic interactions, hydrophobic contacts, π-stacking, and cation-π interactions. The web-server can process large libraries of up to 150'000 docked ligand poses. The results are web-based and can be operated on-line using the built-in HTML5 interactive analysis tools, or can be downloaded for a local use. The vsFilt is freely available on-line, no login required.
Table of contents
[return to toc]
Input
[return to toc]
Docked ligand poses
SDF is a widely used structure-data file format. Associated data items <ID> and <SCORE> could be introduced in the SDF file and used by vsFilt. Ligand IDs should be given in the <ID> item; otherwise, <ID> items will be automatically introduced with a serial ligand number. The <SCORE> item contains any information useful for ligand ranking (e.g., calculated dG); if <SCORE> is introduced, selected ligands will be sorted by the <SCORE> values.
[return to toc]
Protein 3D-model
A protein model containing all (polar and non-polar) hydrogen atoms is required. Note that water molecules should be named HOH, and metal ions - CA2, MG2, or ZN2 in the "residue name" column.
[return to toc]
vsFilt protein-ligand interaction profile
The user has to specify the type of a ligand functional group and the protein residue by which the required interaction should be mediated. "Minimum # of interactions" option allows the identification of ligands whose functional group forms multiple interactions. Available ligand groups are:
| OH | hydroxyl |
| CO | carbonyl |
| COO | carboxyl |
| COOC | ester |
| COC | ether |
| CON | amide |
| NH | amino |
| NAR | N aromatic |
| SO3 | sulfo |
| SO2N | sulfonamide |
| DON | unspecified h-bond donor |
| ACC | unspecified h-bond acceptor |
| HAL | halo involved in halogen bond |
| HPH | involved in hydrophobic interaction |
| STK | involved in stacking interaction |
| PIC | involved in cation-pi interaction |
The interaction profile can be established on-site or uploaded as a text file, and vsFilt then automatically identifies participating atoms and sets criteria for filtering. An alternative interaction profile can be added using the OR operator.
[return to toc]
Apply angle constraints for h-bonds
This option enables angle constraints for H-bonds. All H-bond angles are constrained to be >= 130 degrees.
[return to toc]
Apply tight constraints for h-bonds
This option enables tighter constraints for H-bonds. Maximum H-bond distance: minus 0.5 angstroms. Minimum H-bond angle: plus 20 degrees.
[return to toc]
Output
selected_ligands.txt: the list of selected ligands
selected_ligands.sdf: the selected ligands in SDF format
selected_ligands.tar.gz: PDB files of the selected ligands
vsfilt.log: vsFilt log file
[return to toc]
Demo mode
Demo mode demonstartes the web-server functionality:
the input files are the PARP-1 molecular model (PDB) and poses of 8953 benzamide derivatives docked into its active site (SDF), and the interaction profile is established on-site (alternatively, it can be uploaded as a text file). The "angle constraints" and "tight constraints" are enabled. Here are the input and output files. In this demo, vsFilt selects 17 ligands forming required interactions with the Gly863, Ala898, and Tyr907 residues.
In the SDF file obtained through docking ZINC database compounds (http://zinc.docking.org) with Lead Finder (http://www.biomoltech.com), the ligand IDs are presented in the <ID> item. The <SCORE> item contains VS-scores calculated by Lead Finder for pose ranking and is introduced by renaming the Lead Finder's field <BioMolTech Lead Finder VSscore> to <SCORE>; this enables sorting ligands by the <SCORE> value in the vsFilt output TXT file. The interaction profile contains data for applying structural criteria: it requires two hydrogen bonds with Gly863, hydrophobic contact with Ala863, and stacking interaction with Tyr907. The formation of specified interactions is monitored for each docked ligand, and those ligands that meet the structural criteria are accepted, while the rest are rejected.

❶ The <ID> item in the input SDF file. ❷ The <SCORE> item.

❸ Interaction profile requires two hydrogen bonds with with Gly863.
❹ Hydrophobic contact with the Ala898 side chain.
❺ Stacking with the Tyr907 side chain.
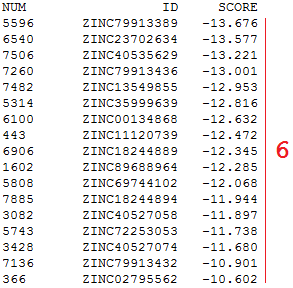
❻ Sorting ligands by the <SCORE> in the output TXT file.
[return to toc]
Citing and contact
Publication: Irina V. Gushchina, Aleksandra M. Polenova, Dmitry A. Suplatov, Vytas K. Švedas, and Dmitry K. Nilov (2020) vsFilt: A Tool to Improve Virtual Screening by Structural Filtration of Docking Poses. J. Chem. Inf. Model. DOI:10.1021/acs.jcim.0c00303
Support: Please send your questions and queries to Dmitry Nilov nilov@belozersky.msu.ru
Links: